A First Introduction to NCBI
David R. Nelson rev. Jan. 5, 2012
The first place to go for access to sequence data is NCBI the National Center for Biotechnology Information. There is no more comprehensive site. If you will be using bioinformatics at all in the future, then you should create a bookmark file for your bioinformatics links so they will be easily accessible. I have set my browser home button to the NCBI site and one of my first bookmarks is to the blast server at NCBI. The link to NCBI is NCBI. For convenience you should make a separate browser window for these notes so you can move back and forth between screens. In Internet Explorer type ctrl+N to make a duplicate. Then minimize the window to store it on the bottom toolbar.
There are many layers and functions at NCBI and we will not go over them all now. To start, I will give you a brief summary of the sequence content in some of the most useful sections of the database. First, go to the
Genbank growth statistics
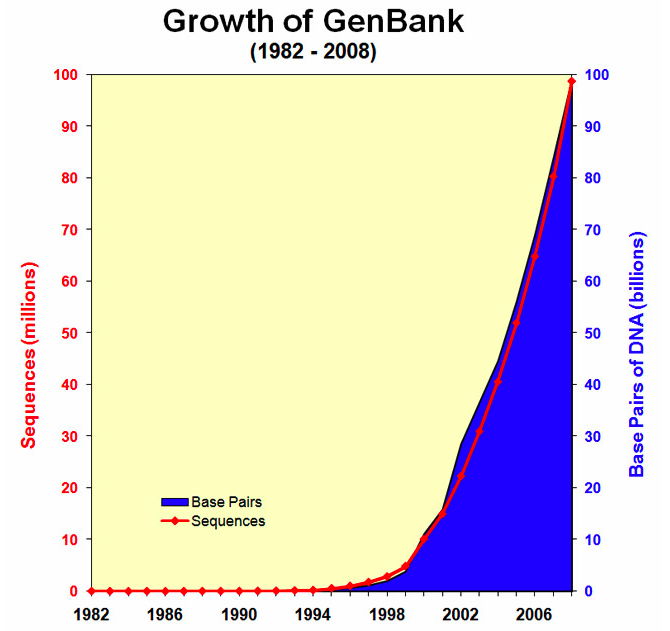
You can see there were about 100 billion nucleotides in 100 million sequences as of Dec. 15, 2008. The growth rate is very steep. As of Dec 15, 2011 there were 135 billion basepairs in 146 million sequences. This figure does not include the Whole Genome Shotgun (WGS) database in the count. The WGS section alone had 241 billion letters in 77 million sequences on Dec. 1, 2011. Total for WGS and Genbank = 376 billion base pairs or Gigabases.
The following tables are taken from the Genbank release notes. These show how sequence data have changed over 2-3 year intervals.
2.2.7 Selected Per-Organism Statistics
The following table provides the number of entries and bases of DNA/RNA for
the most sequenced organisms in Releases: 121.0, Release 139.0, Release 151
and release 163, release 175 release 187 (Dec. 15, 2011). Red species were not in the top in the release beneath it.

The only new species this year compared to 2009 is Solanum lycopersicum (tomato)
The tomato genome has been sequenced and a paper on the genome is currently in
review at Nature.
Release 175 Dec. 15, 2009
Entries Bases Species
13159466 13908099312 Homo sapiens (human)
7860659 8832081330 Mus musculus (mouse
1998941 6279057623 Rattus norvegicus (rat)
2178209 5348086238 Bos taurus (cow)
3889116 5030646878 Zea mays (corn, maize)
2944910 4583294332 Sus scrofa (pig)
1696309 3058371049 Danio rerio (zebrafish)
228206 1352872121 Strongylocentrotus purpuratus (urchin)
1228296 1191090309 Oryza sativa Japonica Group (rice)
1753617 1185599468 Nicotiana tabacum (tobacco)
1423562 1146893384 Xenopus (Silurana) tropicalis (frog)
1204509 1042164589 Drosophila melanogaster (fruitfly)
213485 999212659 Pan troglodytes (chimp)
2283144 991227695 Arabidopsis thaliana (thale cress)
1434990 931947188 Canis lupus familiaris (dog)
655914 911045354 Vitis vinifera (grapevine)
807724 885496928 Gallus gallus (chicken)
1839992 864328945 Glycine max (soybean)
79135 812636075 Macaca mulatta (rhesus monkey)
1118338 793077812 uncultured bacterium
1216178 748172925 Ciona intestinalis (sea squirt)
1139607 697955222 Triticum aestivum (wheat)
1234458 673633441 Oryzias latipes (Medaka, ricefish)
Release 163.0 Dec. 15, 2007
Entries Bases Species
11279087 12854169426 Homo sapiens
7228215 8323105071 Mus musculus
1305109 5778218766 Rattus norvegicus
2880831 4205748958 Zea mays
2057685 3845789573 Bos taurus
1584337 2832324577 Danio rerio
2121316 2304579871 Sus scrofa
227854 1352499678 Strongylocentrotus purpuratus (sea urchin)
1418414 1136849350 Xenopus tropicalis
1130666 1005948286 Oryza sativa Japonica Group
212624 974669347 Pan troglodytes
738789 900252577 Drosophila melanogaster
1951847 881157707 Arabidopsis thaliana
802672 864709966 Gallus gallus
530099 838165327 Vitis vinifera (grapevive)
76348 784577143 Macaca mulatta
1221140 700538475 Canis lupus familiaris
1011784 660118364 Sorghum bicolor
1103658 656437787 Triticum aestivum
422523 536075160 Medicago truncatula
Release 151.0 Dec. 15, 2005 (chloroplast and mitochondrial sequences not included,
and Whole Genome Shotgun sequences not included)
Entries Bases Species
9899176 11953879540 Homo sapiens (human)
6625240 7917536708 Mus musculus (mouse)
1035223 5682651636 Rattus norvegicus (rat)
1356047 2424040780 Bos taurus (cattle)
881280 2215171371 Danio rerio (zebrafish)
2678230 1780527217 Zea mays (maize)
357923 1171720224 Oryza sativa (rice, japonica cultivar-group)
1179107 954916818 Xenopus tropicalis (frog, diploid)
1409440 805877955 Canis familiaris (dog)
534811 785832602 Drosophila melanogaster (fruit fly)
1120673 785475842 Sus scrofa (pig)
787971 684548519 Gallus gallus (chicken)
981930 648235477 Arabidopsis thaliana (thale cress)
207825 533669351 Pan troglodytes (chimp)
784734 463997658 Sorghum bicolor (Sorghum)
68350 434454818 Macaca mulatta (Rhesus monkey)
695995 420688666 Ciona intestinalis (seq squirt)
387953 420276295 Medicago truncatula (barrel medic, plant)
596255 404132383 Brassica oleracea (cabbage, cauliflower, kale)
612712 349010415 Triticum aestivum (wheat)
Release 139.0 December 15, 2003
Whole Genome Shotgun sequences not included
Entries Bases Species
7152768 10307972332 Homo sapiens (human)
5440187 6293834876 Mus musculus (mouse)
843877 5553994470 Rattus norvegicus (rat)
569066 1186984387 Danio rerio (zebrafish)
1700546 1060366537 Zea mays (maize)
266693 714394030 Oryza sativa (rice, japonica cultivar-group)
361257 697671246 Drosophila melanogaster (fruit fly)
887467 508114342 Canis familiaris (dog)
596909 503066134 Gallus gallus (chicken)
589739 424174855 Arabidopsis thaliana (thale cress)
595964 403835636 Brassica oleracea (cabbage, cauliflower, kale)
650813 396135104 Bos taurus (cattle)
175582 385807223 Pan troglodytes (chimpanzee)
25432 337349532 Macaca mulatta (Rhesus monkey)
553918 303123150 Triticum aestivum (wheat)
499249 294204103 Ciona intestinalis (sea squirt, tunicate)
250141 237196637 Medicago truncatula (barrel medic, plant)
222595 233083018 Caenorhabditis elegans (nematode worm)
364210 232049749 Xenopus laevis (African clawed frog, tetraploid)
324322 211144144 Zea mays subsp. Mays (another maize subspecies)
297857 210279451 Silurana tropicalis (another frog)
Release 121.0 December 15, 2000
Entries Bases Species
3918724 6702881570 Homo sapiens (human)
2456194 1291602139 Mus musculus (mouse)
166554 487561384 Drosophila melanogaster (fruit fly)
181388 242674129 Arabidopsis thaliana (thale cress)
114553 203544197 Caenorhabditis elegans (nematode worm)
188993 165539271 Tetraodon nigroviridis (freshwater pufferfish)
151411 125948974 Oryza sativa (rice)
218598 106344366 Rattus norvegicus (rat)
159473 71215626 Bos taurus (cattle)
141802 62817102 Glycine max (soybeans)
104535 50991920 Medicago truncatula (barrel medic, plant)
91334 49855996 Trypanosoma brucei (protozoan)
97112 49415566 Lycopersicon esculentum (tomato)
54328 47639714 Giardia intestinalis (hikers Diarrhea protist)
77532 47590936 Strongylocentrotus purpuratus (sea urchin)
49938 44522016 Entamoeba histolytica (parasite)
57779 44489692 Hordeum vulgare (barley)
83726 40906902 Danio rerio (zebrafish)
77506 36885212 Zea mays (maize)
18361 32779082 Saccharomyces cerevisiae (bakers yeast)
These sequences are in several sections and it is important to know about these so you can find what you are looking for at Genbank. This table shows the breakdown of the four main sequence divisions in Genbank in Jan 2010. For 2004 data see this older table. These are nr (non-redundant), est for expressed sequence tags, htgs for high throughput genomic sequences and gss for genome survey sequences. STS is a small section called sequence tagged sites used in mapping. PAT is a patent section. We will talk about each of them in more detail. Nr is the default section of the database that you will search when you do a BLAST search unless you select another section. It is a good place to start. It contains all the genes that have been sequenced by individuals over the years, It has both cDNA and gene sequences. Genome project sequences move into nr as they are finished. Unless a submitter is sending in a batch submission of large numbers of sequences for the other sections of the database their sequence goes here. Note that as of Dec. 1, 2011 nr has 34.8 billion bases or 25% of the total Genbank bases. If you only search here for a match to your sequence, you are neglecting 75% of Genbank data.
Est is the division for sequencing projects focusing on cDNAs, and usually large numbers of ESTs are deposited from these projects. I break down the human, mouse and other ests to show that 20% are human or mouse and 80% are other. Note, there are more bases in the EST section than in the nr section. The HTGS section is for genomic sequencing. This is where the genome projects used to send their data. It was the largest section of Genbank in 2004, but now this genome sequence data is going to the WGS database. HTGS has a lot of human and mouse sequence.
GSS stands for genome survey sequence. Before Next Gen sequencing (Illumina, etc.) most genome projects were based on sequencing clones of differing sizes. Some are cosmids and some are BACs or Bacterial Artificial Chromosomes. These are too big to sequence in one run so they need to be broken into smaller pieces. One way to keep track of BACs is to end sequence them to get a short sequence marker for mapping and later assembly. The BAC end sequences go in the GSS section of Genbank. They are similar in size to the ESTs but they are from genomic DNA. There are ~21 billion bases of GSS sequence. I recommend searching all four main sections for matches to any sequence you have, since you don't want to miss anything. The smaller section called STS (only 1.3 million sequences) contains sequences used for mapping. I rarely search STS anymore since the sequence it contains is mostly covered in other sections.
The NCBI Sitemap has links to all the different parts of NCBI, including the dbEST, WGS, trace archive and Blast pages.
Follow the alphabet bar to D for databases. dbEST and dbGSS are linked from this table. Click on the dbEST link to find info on this database. dbEST summary by organism lists the most sequenced organisms in dbEST. Below
I have copied the entries with the most ESTs from 2004, 2005, 2007, 2009 and 2011. ESTs are sequence fragments derived from cDNAs, so they represent genes that are expressed as mRNA.

dbEST release 123109
Summary by Organism - December 31, 2009
Number of public entries: 64,526,769
link to data
Homo sapiens (human) 8,296,280
Mus musculus + domesticus (mouse) 4,852,144
Zea mays (maize) 2,018,798
Bos taurus (cattle) 1,558,493
Sus scrofa (pig) 1,538,441
Arabidopsis thaliana (thale cress) 1,527,298
Danio rerio (zebrafish) 1,481,930
Glycine max (soybean) 1,422,982
Xenopus (Silurana) tropicalis (western clawed frog) 1,271,375
Oryza sativa (rice) 1,249,110
Ciona intestinalis 1,205,674
Triticum aestivum (wheat) 1,067,291
Rattus norvegicus + sp. (rat) 1,009,817
Drosophila melanogaster (fruit fly) 821,005
Xenopus laevis (African clawed frog) 677,806
Oryzias latipes (Japanese medaka) 665,382
Brassica napus (oilseed rape) 643,601
Gallus gallus (chicken) 600,323
Hordeum vulgare + subsp. vulgare (barley) 501,614
Salmo salar (Atlantic salmon) 494,392
Panicum virgatum (switchgrass) 442,269
Phaseolus coccineus 391,138
Canis lupus familiaris (dog) 365,909
Physcomitrella patens subsp. patens 362,131
Vitis vinifera (wine grape) 357,856
Caenorhabditis elegans (nematode) 355,321
Ictalurus punctatus (channel catfish) 354,434
dbEST release 121407
Summary by Organism - December 14, 2007
Homo sapiens (human) 8,135,176
Mus musculus + domesticus (mouse) 4,850,243
Bos taurus (cattle) 1,497,772
Sus scrofa (pig) 1,475,731
Danio rerio (zebrafish) 1,379,829
Arabidopsis thaliana (thale cress) 1,276,695
Xenopus tropicalis (western clawed frog) 1,271,375
Oryza sativa (rice) 1,214,088
Zea mays (maize) 1,174,690
Triticum aestivum (wheat) 1,051,175
Rattus norvegicus + sp. (rat) 895,827
Ciona intestinalis 686,396
Xenopus laevis (African clawed frog) 677,784
Gallus gallus (chicken) 599,330
Brassica napus (oilseed rape) 567,177
Drosophila melanogaster (fruit fly) 542,677
Hordeum vulgare + subsp. vulgare (barley) 467,916
Salmo salar (Atlantic salmon) 432,815
Glycine max (soybean) 392,393
Canis familiaris (dog) 365,909
Vitis vinifera (wine grape) 352,984
Caenorhabditis elegans (nematode) 346,107
Oryzias latipes (Japanese medaka) 343,846
Pinus taeda (loblolly pine) 328,628
Physcomitrella patens subsp. patens 305,606
Aedes aegypti (yellow fever mosquito) 298,060
Branchiostoma floridae (Florida lancelet) 277,538
Gasterosteus aculeatus (three spined stickleback) 276,992
Picea glauca (white spruce) 272,464
Oncorhynchus mykiss (rainbow trout) 260,886
Solanum lycopersicum (tomato) 257,540
Malus x domestica (apple tree) 255,111
Pimephales promelas (fish) 249,941
Medicago truncatula (barrel medic) 249,450
Saccharum officinarum (sugarcane) 246,301
Solanum tuberosum (potato) 230,780
Sorghum bicolor (sorghum) 209,776
Chlamydomonas reinhardtii 202,044
Ixodes scapularis (black-legged tick) 193,480
Ovis aries (sheep) 186,678
Bombyx mori (domestic silkworm) 184,509
Aplysia californica (California sea hare) 179,000
Gossypium hirsutum (upland cotton) 177,244
Hydra magnipapillata (pond hydra) 174,162
Nematostella vectensis (sea anemone) 162,714
Schistosoma mansoni (blood fluke) 158,841
Nicotiana tabacum (tobacco) 158,008
Dictyostelium discoideum (cellular slime mold) 155,032
Anopheles gambiae (African malaria mosquito) 153,165
Lotus japonicus (plant) 150,631
Brassica rapa subsp. pekinensis (Chinese cabbage) 147,217
Trichosurus vulpecula (brushtail possum) 147,199
Strongylocentrotus purpuratus (purple urchin) 141,833
Paracentrotus lividus (sea urchin) 140,897
Picea sitchensis (Sitka spruce) 139,569
Capitella sp. I ECS-2004 (marine polychaete worm) 138,404
Toxoplasma gondii (parasite, pathogen) 129,421
Gadus morhua (Atlantic cod) 126,325
Acyrthosiphon pisum (pea aphid) 120,255
Molgula tectiformis (sea squirt, ascidian) 106,863
Helobdella robusta (leech, annelid worm) 101,359
Macaca fascicularis (cynomolgus monkey) 101,192
Summary by Organism - Nov. 11, 2005
Entries with more than 100,000 ESTs
Number of public entries: 31,307,034
Homo sapiens (human) 7,057,754
Mus musculus + domesticus (mouse) 4,688,047
Xenopus tropicalis (frog) 1,038,272
Rattus sp. (rat) 704,494
Bos taurus (cattle) 702,645
Danio rerio (zebrafish) 689,581
Ciona intestinalis (sea squirt, a tunicate) 686,396
Zea mays (maize) 656,945
Triticum aestivum (wheat) 600,039
Gallus gallus (chicken) 578,445
Sus scrofa (pig) 502,501
Xenopus laevis (African clawed frog) 473,792
Arabidopsis thaliana (thale cress) 420,789
Oryza sativa (rice) 406,790
Hordeum vulgare + subsp. vulgare (barley) 395,019
Drosophila melanogaster (fruit fly) 383,407
Glycine max (soybean) 355,978
Canis familiaris (dog) 349,306
Pinus taeda (loblolly pine) 329,469
Caenorhabditis elegans (nematode, round worm) 302,080
Branchiostoma floridae (Florida lancelet, Amphioxus) 277,538
Pimephales promelas (minnow) 249,938
Saccharum officinarum (sugar cane) 246,301
Oncorhynchus mykiss (rainbow trout) 239,327
Oryzias latipes (Japanese medaka) 221,546
Solanum tuberosum (potato) 219,765
Medicago truncatula (barrel medic) 216,703
Aedes aegypti (yellow fever mosquito) 213,805
Sorghum bicolor (sorghum) 208,466
Lycopersicon esculentum (tomato) 199,279
Malus x domestica (apple tree) 197,774
Vitis vinifera (grapes) 190,434
Hydra magnipapillata (hydra, radial animal) 174,162
Gasterosteus aculeatus (stickleback fish) 170,994
Chlamydomonas reinhardtii (green alga) 167,641
Schistosoma mansoni (blood fluke) 158,841
Dictyostelium discoideum (cellular slime mold) 155,032
Anopheles gambiae (African malaria mosquito) 153,165
Strongylocentrotus purpuratus (purple sea urchin) 130,988
Bombyx mori (domestic silkworm) 127,748
Toxoplasma gondii (protozoan parasite) 125,741
Physcomitrella patens subsp. patens (moss) 120,702
Salmo salar (Atlantic salmon) 113,002
Lotus corniculatus (Birdsfoot trefoil plant, legume) 111,623
Molgula tectiformis (ascidian, tunicate, like Ciona) 106,863
Picea glauca (white spruce) 104,305
Summary by Organism - January 2, 2004
Number of public entries: 19,635,256
Homo sapiens (human genome sequenced) 5,469,433
Mus musculus + domesticus (mouse genome sequenced) 4,030,839
Rattus sp. (rat genome almost sequenced) 558,402
Triticum aestivum (wheat) 549,915
Ciona intestinalis (sea squirt, a urochordate, genome seq)492,511
Gallus gallus (chicken) 451,655
Danio rerio (zebrafish) 405,962
Zea mays (maize) 391,145
Xenopus laevis (African clawed frog, tetraploid) 357,038
Hordeum vulgare + subsp. vulgare (barley) 348,282
Glycine max (soybean) 344,524
Bos taurus (cattle) 331,139
Silurana tropicalis (frog, diploid smaller genome) 297,086
Drosophila melanogaster (fruit fly genome sequenced) 267,332
Oryza sativa (rice genome sequenced) 266,949
Saccharum officinarum (sugar cane) 246,301
Sus scrofa (pig genome is being sequenced) 240,001
Caenorhabditis elegans (nematode worm, genome sequenced) 215,200
Arabidopsis thaliana (thale cress genome sequenced) 196,904
Medicago truncatula (barrel medic) 187,763
Sorghum bicolor (sorghum a grass) 161,766
Dictyostelium discoideum (slime mold genome sequenced) 155,032
Chlamydomonas reinhardtii(green algae,genome sequenced) 154,600
Lycopersicon esculentum (tomato) 150,410
Schistosoma mansoni (blood fluke) 139,135
Oncorhynchus mykiss (rainbow trout) 137,127
Vitis vinifera (grape) 135,712
Anopheles gambiae (African malaria mosquito genome seq) 134,784
Solanum tuberosum (potato) 132,122
Pinus taeda (loblolly pine) 110,622
Oryzias latipes (Japanese medaka) 103,098
Physcomitrella patens subsp. patens (moss) 82,313
Toxoplasma gondii (protozoan parasite) 72,859
Lactuca sativa (garden lettuce) 68,188
Populus tremula x Populus tremuloides (aspen tree hybrid) 65,981
Helianthus annuus (sunflower) 59,841
Salmo salar (salmon) 59,420
Strongylocentrotus purpuratus (purple sea urchin) 51,744
Notice that mouse and human have over 13 million ESTs between them. Note the red line is Zea mays (maize). We will use these ESTs in our assignment today.
dbGSS summary by organism lists the most sequenced organisms in dbGSS. Below
I have copied the entries with the most GSS sequences from 2004, 2005, 2007, 2009 and 2011.
dbGSS release 123109
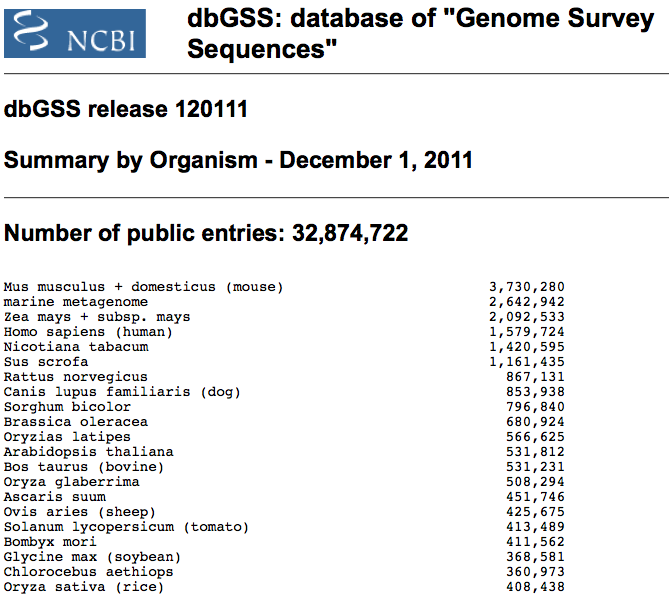
dbGSS release 123109
Summary by Organism - December 31, 2009
Number of public entries: 27,301,327
marine metagenome 2,642,942
Mus musculus + domesticus (mouse) 2,437,343
Zea mays + subsp. mays (maize) 2,091,983
Nicotiana tabacum (tobacco) 1,420,579
Homo sapiens (human) 1,268,770
Sus scrofa (pig) 981,378
Rattus norvegicus (rat) 866,532
Canis lupus familiaris (dog) 853,938
Sorghum bicolor 796,840
Brassica oleracea 680,836
Oryzias latipes (ricefish) 566,625
Bos taurus (bovine) 515,056
Arabidopsis thaliana 509,506
Ascaris suum 451,746
Ovis aries (sheep) 425,689
Bombyx mori (silkworm) 411,562
Glycine max (soybean) 368,551
Chlorocebus aethiops (African green monkey) 360,973
Solanum phureja (a kind of potato) 343,872
Mus musculus molossinus 337,492
Solanum lycopersicum (tomato) 319,766
Equus caballus (horse) 315,533
Oryza sativa (rice) 299,369
Summary by Organism - December 14, 2007
marine metagenome 2,642,942
Zea mays + subsp. mays 2,083,122
Mus musculus 1,845,032
Homo sapiens (human) 1,211,538
Canis familiaris (dog) 853,938
Sorghum bicolor 794,962
Brassica oleracea 595,474
Sus scrofa 595,150
Oryzias latipes 566,625
Bos taurus (bovine) 514,766
Arabidopsis thaliana 488,181
Ascaris suum 451,746
Ovis aries (sheep) 425,679
Glycine max (soybean) 368,246
Mus musculus molossinus 337,492
Solanum lycopersicum (tomato) 319,461
Equus caballus 315,464
Rattus norvegicus 307,774
Oryza sativa (rice) 280,447
Oryza australiensis 271,727
Chlorocebus aethiops 253,760
Oryza ridleyi 215,820
Oryza coarctata 204,809
Pan troglodytes 197,579
Ustilago maydis 193,251
Tetraodon nigroviridis 188,963
Brassica rapa subsp. pekinensis 179,678
Oryza minuta 179,008
Macropus eugenii 178,807
Medicago truncatula (barrel medic) 168,815
Gallus gallus (chicken) 164,629
Danio rerio 161,275
Oryza granulata 144,859
Oryza rufipogon 143,074
Solanum tuberosum 141,471
Oryza alta 136,269
Xenopus tropicalis 131,325
Aedes aegypti (yellow fever mosquito) 118,058
Oryza nivara 109,315
Vitis vinifera 109,147
Oryza officinalis 105,939
Drosophila melanogaster 102,113
dbGSS release 111105
Summary by Organism - November 11, 2005
Number of public entries: 13,340,273
Entries with more than 100,000 GSS reads
Zea mays + subsp. mays 1,942,149
Mus musculus 1,515,692
Homo sapiens (human) 936,533
Canis familiaris (dog) 853,905
Brassica oleracea 595,420
Sorghum bicolor 573,724
Bos taurus (bovine) 488,965
Arabidopsis thaliana 416,123
Ovis aries (sheep) 376,602
Mus musculus molossinus 337,492
Sus scrofa 589,992
Rattus norvegicus 307,774
Oryza sativa (rice) 242,464
Oryza coarctata 195,285
Tetraodon nigroviridis 188,963
Lycopersicon esculentum (tomato) 184,832
Pan troglodytes 176,409 Oryza minuta 169,651
Medicago truncatula (barrel medic) 168,679
Gallus gallus (chicken) 164,557
Danio rerio 159,832
Oryza australiensis 137,530
Oryzias latipes 135,781
Xenopus tropicalis 131,325
Oryza alta 128,732
Aedes aegypti (yellow fever mosquito) 118,056
Oryza nivara 106,124
Oryza officinalis 103,251
dbGSS release 010204
Summary by Organism - January 2, 2004
Number of public entries: 7,968,534
Zea mays + subsp. mays (maize, corn) 1,618,438
Mus musculus (mouse) 1,156,190
Homo sapiens (human) 896,054
Canis familiaris (dog) 853,888
Brassica oleracea (cabbage, cauliflower) 595,321
Bos taurus (cattle) 309,619
Rattus norvegicus (rat) 307,593
Arabidopsis thaliana (thale cress) 286,669
Tetraodon nigroviridis (pufferfish) 188,963
Danio rerio (zebrafish) 159,024
Pan troglodytes (chimpanzee) 158,594
Gallus gallus (chicken) 137,133
Aedes aegypti (yellow fever mosquito) 118,044
Oryza sativa (rice) 108,060
Trypanosoma brucei (parasite) 90,847
Entamoeba histolytica (parasite) 79,674
Strongylocentrotus purpuratus(sea urchin) 76,019
Xenopus tropicalis (frog) 66,547
Medicago truncatula (barrel medic) 61,232
Anopheles gambiae (mosquito) 60,352
Drosophila melanogaster (fruit fly) 56,954
Takifugu rubripes (Japanese pufferfish) 50,808
GSS is a database of genomic fragments about the same size as ESTs, but these of course contain introns and non-coding regions as well as exons. Please note the organisms represented are different than the ESTs. This can be of help sometimes. Notice there are 10 different species of rice (Oryza). Oryzias latipes is not rice, but a small fish that grows in the rice paddies. Oryzias, Tetraodon nigroviridis, the freshwater pufferfish and Danio (zebrafish) are all fish. These genes can be surprisingly similar to human and they are helpful when trying to assemble genes from genomic DNA when you cannot find the intron-exon boundaries. Do not forget them, they can help you.
Let's look more closely at the nr section. Release notes for GenBank show the following. The Genbank release is broken into several hundred files. The Genbank sequences are categorized into the following 19 divisions. If the EST, GSS, STS, PAT, HTG, CON and ENV files are ignored, nr is what's left. That is the sum of divisions 1-11 + 17.
The newest data on divisions from EMBL, Dec. 1, 2011. EMBL has some differences
from Genbank. Fungi is broken out from Plants, there is no Primate section, etc.
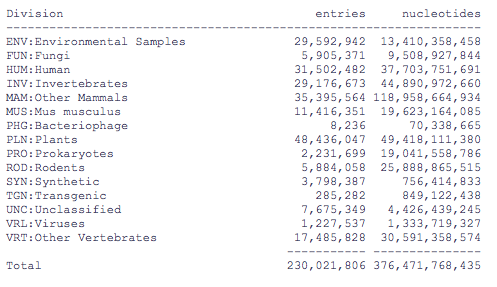
Data from Jan 6, 2010
1. PRI - primate sequences 11,461,667
2. ROD - rodent sequences 3,916,991
3. MAM - other mammalian sequences 14,250,223
4. VRT - other vertebrate sequences 2,856,393
5. INV - invertebrate sequences 6,550,758
6. PLN - plant, fungal, and protist sequences 3,681,540
7. BCT - bacterial sequences (includes Archaea) 1,485,534
8. VRL - viral sequences 795,464
9. PHG - bacteriophage sequences 5,952
10. SYN - synthetic sequences 91,574
11. UNA - unannotated sequences 225
12. EST - EST sequences (expressed sequence tags) 64,569,628
13. PAT - patent sequences 12,388,931
14. STS - STS sequences (sequence tagged sites) 1,311,239
15. GSS - GSS sequences (genome survey sequences) 27,049,249
16. HTG - HTG sequences (high-throughput genomic sequences) 143,922
17. HTC - unfinished high-throughput cDNA sequencing 551,480
18. CON - constructed (for details on assembly of contigs) 7,648,171
19. ENV - environmental samples 19,702,621
Data from Jan 9, 2008
1. PRI - primate sequences 4,286,364
2. ROD - rodent sequences 3,851,113
3. MAM - other mammalian sequences 9,825,135
4. VRT - other vertebrate sequences 1,810.578
5. INV - invertebrate sequences 2,589,791
6. PLN - plant, fungal, and protist sequences 1,369,982
7. BCT - bacterial sequences (includes Archaea) 450,292
8. VRL - viral sequences 524,240
9. PHG - bacteriophage sequences 3,741
10. SYN - synthetic sequences 55,697
11. UNA - unannotated sequences 4,124,711
12. EST - EST sequences (expressed sequence tags) 49,040,258
13. PAT - patent sequences 4,745,089
14. STS - STS sequences (sequence tagged sites) 933,316
15. GSS - GSS sequences (genome survey sequences) 21,371,486
16. HTG - HTG sequences (high-throughput genomic sequences) 119,165
17. HTC - unfinished high-throughput cDNA sequencing 493,976
18. CON - constructed (for details on assembly of contigs) 4,228,826
19. ENV - environmental samples 2,044,479
Data from Jan 11, 2006
1. PRI - primate sequences 1,962,405
2. ROD - rodent sequences 2,640,641
3. MAM - other mammalian sequences 5,575,208
4. VRT - other vertebrate sequences 532,022
5. INV - invertebrate sequences 1,428,656
6. PLN - plant, fungal, and protist sequences 735,872
7. BCT - bacterial sequences (includes Archaea) 261,480
8. VRL - viral sequences 331,748
9. PHG - bacteriophage sequences 2,886
10. SYN - synthetic sequences 25,747
11. UNA - unannotated sequences 1,037,720
12. EST - EST sequences (expressed sequence tags) 32,633,559
13. PAT - patent sequences 2,685,324
14. STS - STS sequences (sequence tagged sites) 880,049
15. GSS - GSS sequences (genome survey sequences) 13,671,106
16. HTG - HTG sequences (high-throughput genomic sequences) 80,350
17. HTC - unfinished high-throughput cDNA sequencing 435,673
18. CON - contig (for details on assembly of contigs) 309,981
19. ENV - environmental samples 206,786
Data from April 15 2001
1. PRI - primate sequences 1,111,961
2. ROD - rodent sequences 484,969
3. MAM - other mammalian sequences 1,146,543
4. VRT - other vertebrate sequences 143,570
5. INV - invertebrate sequences 375,247
6. PLN - plant, fungal, and protist sequences 530,861
7. BCT - bacterial sequences (includes Archaea) 239,646
8. VRL - viral sequences 214,974
9. PHG - bacteriophage sequences 2,481
10. SYN - synthetic sequences 9,793
11. UNA - unannotated sequences 1,146
12. EST - EST sequences (expressed sequence tags) 19,667,401
13. PAT - patent sequences 1,489,482
14. STS - STS sequences (sequence tagged sites) 258,123
15. GSS - GSS sequences (genome survey sequences) 8,109,223
16. HTG - HTG sequences (high-throughput genomic sequences) 67,813
17. HTC - unfinished high-throughput cDNA sequencing 149,393
18. CON - contig (for details on assembly of contigs) 3,639
note: these numbers can be found by Entrez nucleotide search for gbdiv_pri etc.
These three letter designations appear in the top line of any sequence you pull out of Genbank. EST has the largest number of entries. GSS is almost half as large. Mammals (first three divisions) are heavily sampled. Other vertebrate (zebrafish, xenopus etc) is quite a bit smaller. Invertebrate is pretty large since it has both fly and C. elegans sequences in it. Plant is a catch-all section that has every eukaryotic species that is not an animal. This includes fungi and protists, even though they are not plants. Bacteria has all the public bacterial genomes including Archaea. 2668 bacterial genomes and 169 archael genomes are now sequenced. Viral rounds out the nr section. Many complete viral genomes have been sequenced. The genomes section of Genbank, Entrez Genomes currently contains 3962 Reference Sequences for 2749 viral genomes as of Jan. 5, 2012. (http://www.ncbi.nlm.nih.gov/genomes/VIRUSES/viruses.html)
You can specify many of these categories except invertebrate when doing a blast search to limit your search. This can be done using the organism window in the middle of the blast page. The first letters you type bring up an automatic menu of choices that match that spelling. If you type in fu, fungi appears. You usually do not need to type in more than 2-4 characters to find your choice. As I mentioned above, invertebrates does not work since invertebrates is not a recognized taxonomic choice in this menu. To limit your search to invertebrates you can use the Entrez query window below the organism window. You must use proper Entrez syntax to do this and there is a help button to guide you. Metazoa[orgn] NOT vertebrata[orgn] in this window will limit the search to invertebrates. You need the [orgn] tag to make this work. Another example would be Arthropoda[orgn] NOT insects[orgn]. This will find crabs, shrimp, other crustaceans like Daphnia, ticks and other chelicerates but it will exclude insects.
There is one more section of Genbank not included in the 19 divisions listed above. This is the WGS section (for Whole Genome Shotgun sequences). This section is on the pull down menu with NR, GSS, EST and others. It contains 42 million sequences and 156 billion letters (Jan. 6, 2010). These are from 1954 different genome projects including 1153 species, 601 different genera and 134 metagenome projects.
One other source of sequence data is the trace archive at NCBI (and EBI). This
archive is a repository for the raw sequence reads from large sequencing projects.
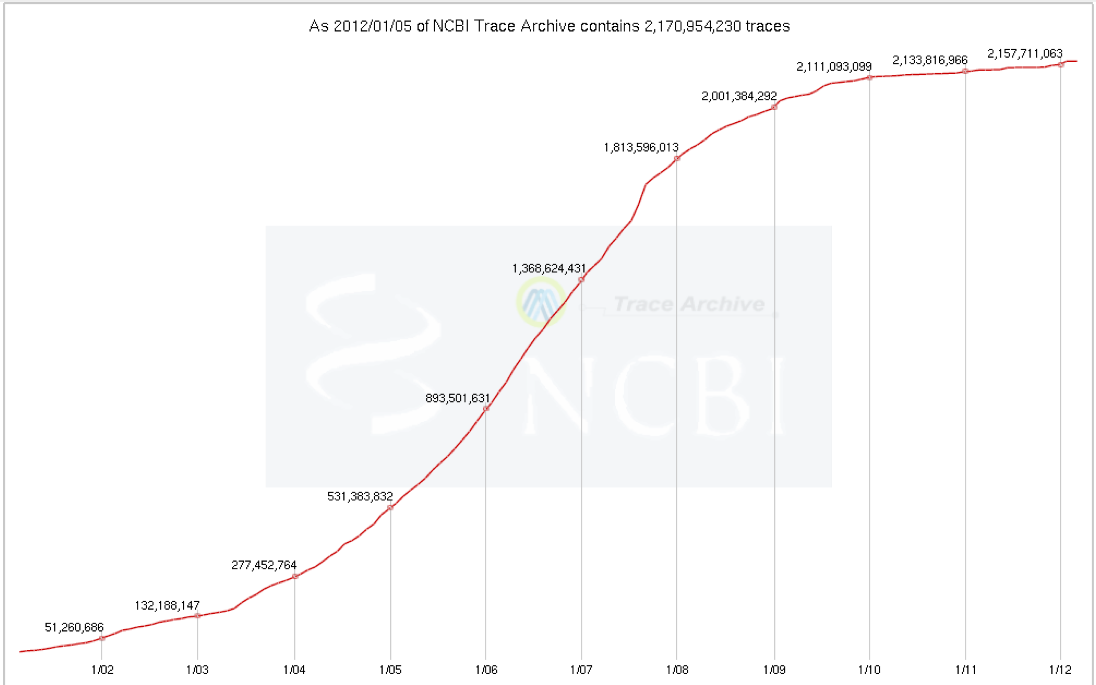
These are Sanger sequencing reads as opposed to 454 and other Next Gen sequencing platforms. There are many copies of each region reflecting the fold coverage of the sequencing project. The graph below shows the growth of the Trace Archive.
2.1 billion traces is about 2.1 trillion bases. That is ~6 times the
size of Genbank and WGS added together. The Trace archive is more difficult to
use, since it can only be searched by nucleotide searches using Megablast and the
pieces are smaller than whole genes, so they are disconnected fragments. This
makes it very difficult to assemble an intact gene.
The newest sequencing methods make terabytes of data from millions of very short fragments (~75-100 bp reads). Because they are so short, higher coverage is needed to assemble an accurate sequence, usually 35-200X as compared to 8-10X for Sanger reads of about 1000 bp. There is a new repository for these sequence data called SRA or short read archive. The data is stored by project and may include up to 16 terabytes in a single project. This data is not searchable by BLAST.
Blast Searching at NCBI
That gives you a flavor of what is in Genbank. Now how do you search this data? The method is the BLAST search. Please go to the NCBI homepage.
On the right side under Popular Resources there is a link to the BLAST page. Go there now.

You will notice that there are several types of blast search methods depending
on what you have and what you want to find. Notice the five sections under Basic
BLAST on this page. Nucleotide blast searches, Protein blast searches and the
translated blast searches: blastx, tblastn and tblastx. If you have nucleotide sequence that is from an untranslated
region, or you do not know what coding sequence is in it, the nucleotide blast
blastn is what you should try to identify your sequence. It may match a known
gene. If you have protein sequence and you want to search the known protein
translations in Genbank blastp is one of your choices. The translated blast
searches are more powerful and they take more computer time. Tblastn is one of
the most useful search tools. This is the one I use almost all of the time with
fragments of genes that I have been able to translate into probable open
reading frames based on other known protein sequences. With tblastn you can
search your protein sequence against all the nucleotides in Genbank sections
that are translated in all six reading frames (three per DNA strand). You can
hardly miss anything that matches if it is out there. Nucleotide searches are
not as sensitive because there are only four bases but there are 20 amino
acids, so there are better search statistics to find a match if you use protein
sequence. The blastx program lets you find ORFs (open reading frames) in your
nucleotide sequence by hunting for matches to translated proteins. This is very
helpful in analyzing fresh genomic sequence that is unannotated. The tblastx
program is the most computer intensive since it does the translation of the
query sequence in six frames as well as the translation of the nulceotide
database in 6 frames, so it is like doing 6 tblastn searches. Many local
sequence database servers will not offer this search, since it eats computer
time so badly.
Blast searches require that you paste or upload a sequence to the program
window as shown below.

This is often just raw sequence, but sometimes the program asks for FASTA
format. FASTA format always starts with an >IDENTIFIER LINE This may be
anything you want to identify your sequence. This will appear in the search
output. If you will be doing many searches and saving them or printing them, it
is a good idea to include this identifier line so you will not mix up your
output. The > symbol is required, otherwise the program will try to
interpret your identifier as amino acid or nucleotide sequence. The sequence
can have numbers in it such as are found in the Genbank sequence format, or
other formats like GCG or PIR. These will be ignored. Non-standard letters like
O, and J will also be ignored. This is an example of FASTA format.
>Dictyostelium CYP51 P450 C-terminal
ETQKDINDIVQKENQGEINFDGLKRMNRLETVIREVLRLHPPLIFLMRK
VMTPMEYKGKTIPAGHILAVSPQVGMRLPTVYKNPDSFEPKRFDVED
KTPFSFIAFGGGKHGCPGENFGILQIKTIWTVLSTKYNLEVGPVPPTD
FTSLVAGPKGPCMVKYSKKQK*
Once your sequence is pasted in the window, you need to select a database from a pull down menu. Nr/nt is the default. Below the Choose Search Set section
is the blast button. Do not click this yet. Underneath the BLAST button is algorithm parameters. These includes the filter option. I usually recommend turning the filter off, unless
there is a weird repetive sequence like a run of QQQQQ in your sequence. If you
leave the filter on, you may find some unpleasant runs of XXXXXXX in the blast
output where you do not want it. One of the best features of this blast
page is the limitation of sequences to be searched. This is found in the Choose Search Set section. Here you can type in a species like
mouse (Mus musculus) or a whole taxonomic range of
organisms like green plants (Viridiplantae) or Fungi. (All organisms is the
default). You can also choose to type in a very specific set yourself like
Diplomonadida or Rhodophyta. The program follows your spelling and offers you the closest options so you often do not have to type in more than three letters. In the Entrez Query window you can enter boolean queries such as metazoa[orgn] NOT vertebrata[orgn]. This will give you invertebrate sequences.
I use this feature all the time to restrict the search output to just what I
want. For the Dictyostelium test sequence above, you may select Dictyostelium discoideum from the organism menu if you
want to find the exact sequence above, or you may expand the search to closely
related organisms by typing in Mycetozoa. It may be more interesting to use
Fungi on this first blast, since there are many CYP51s
known from fungi in the nr database.
Once you press the BLAST button, You will get a screen that refreshes every few seconds until the search is done. This final output will give you
another window with a graphical box at the top that has
colored bars representing the hits. Red is for the highest scoring hit and the
colors get cooler as the scores drop. Red hits are generally good matches.
Since we searched a slime mold vs. Fungi here, the best hits are magenta not
red.

Below the graphics box is a text list of your hits with the best scores at the
top. The E value numbers (6e-20 etc.) refer to the chance that this was an
accidental match. The larger the negative exponent the better the match and the
lower the probability that it was a chance occurrence.

This list is hyperlinked to the actual Genbank entries for each sequence. Below
the text list are the actual alignments. This is where you see what you have
found.

The output here shows a gap in the Dicty sequence compared to most of the
fungal sequences. Since fungi have relatively few introns, this is probably not
an intron in the fungi, but it may be an insertion that is found in fungi and
not in the slime mold sequence. Notice that all the top hits are identified as
CYP51 sequences or 14 alpha demethylase enzymes. This is because the cytochrome
P450 family is extensively annotated and more than 16,000 of the genes are named.
Even the new sequences that are not officially named are usually identified by
this type of blast search and the authors often tag them as a P450 (in this
case CYP51).
Your first search was intended to get you started in blast searching with a simple example. There are many aspects of this tool that we need to discuss in more detail. The first is the selection of the different databases to search. Most first time users of a software package tend to use the default settings. In this case that could be disasterous, because you could miss a large number of sequences that are not to be found in the nr section of Genbank (the default). There are large EST projects on some organisms that will make only EST sequences. These will not be found in nr. Two examples are Triticum aestivum (wheat) and Zea mays (maize or corn) with >1,000,000 EST sequences each. If you are after a plant gene, it may be present in these ESTs but not in nr. The 1,500,000 Bos taurus (cattle) ESTs can also be used to help assemble human, mouse or other vertebrate genes, by indicating the location of introns. On human chromosome 6 There is a gene with an intron that is 479kb long. This gene could not have been assembled without an mRNA or EST sequence to show that the first exon was that far from the rest of the gene. Another example is Giardia (hiker’s diarrhea). Giardia is a common human pathogen and it is thought to be a very ancient eukaryote. Therefore, it is valuable to sequence for medical and evolutionary studies. The Giardia Genome project is being conducted at Woods Hole Oceanographic Institute and the data is searchable at NCBI in the HTGS section of Genbank. (HTGS = High Throughput Genomic Sequences).
If you have not added the blast page at NCBI to your bioinformatics bookmark file, please do it now. NCBI Blast page
I recommend that you add the TBLASTN page also. TBLASTN
You will be needing these over and over as we progress. As a general rule, it is good policy to search nr, HTGS, EST and GSS sections of Genbank when hunting for matches to a new sequence. If your species is in WGS you will definitely want to look there also.
The following is a real example taken from an alignment of mitochondrial carrier sequences. Mitochondrial carriers are membrane transport proteins of the inner mitochondrial membrane (and peroxisomal and hydrogenosomal membranes). These carriers enable communication betweeen the mitochondrial matrix and the cytosol. They are responsible for exporting ATP to the cytosol and importing ADP into the matrix to be remade into ATP. There are 46 of these genes in humans and similar numbers in other species (35 in yeast). I am posting a link to a sequence alignment of 265 of these carriers that has some gaps in some of the sequences. For demonstration purposes we will now try to fill in some of these gaps. Once you are at the page use the find command to search for GMPA-. This should take you to a small gap in sequence 92. This gap was not possible to fill when the alignment was made, because this sequence did not exist in Genbank. The sequence looks like this with eight missing amino acids.
90 -------------ADENGRVGGINLLTAGALAGVPAASLVTPADVIKTRLQVAARAGQ-----TTYSGVV
91 -------------ANEDGQVSPGSLLLAGAIAGMPAASVVTP-DVIKTRLQVAARAGQ-----TTYSGVI
92 -------------ANEDGQVSPGSLLLAGAIAGMPA--------VIKTRLQVAARAGQ-----TTYNGVT
92A ------------PSPEDKTYNILRKVIIAGLASSLACVMSVTLDMAKCRIQGPQPVKG----EVKYQWTI
The second gap is an alignment gap and there are no missing amino acids here. Copy the sequence 92 above and paste it in the blast window at NCBI. Leave the dashes in the sequence. Select the EST database and turn the filter off. To reduce searching unwanted sequences type in mouse in the organism window. For this search limit to mouse ESTs. Do the blast.
>gi|1387895|gb|W76821.1|W76821 me73f10.r1 Soares mouse embryo NbME13.5 14.5 Mus musculus cDNA
clone IMAGE:401227 5' similar to WP:K02F3.2 CE01348 ;.
Length = 455
Score = 48.1 bits (113), Expect = 2e-05
Identities = 23/23 (100%), Positives = 23/23 (100%)
Frame = +3
Query: 1 ANEDGQVSPGSLLLAGAIAGMPA 23
ANEDGQVSPGSLLLAGAIAGMPA
Sbjct: 384 ANEDGQVSPGSLLLAGAIAGMPA 452
>gi|1387483|gb|W77458.1|W77458 me66c04.r1 Soares mouse embryo NbME13.5 14.5 Mus musculus cDNA
clone IMAGE:400518 5' similar to WP:K02F3.2 CE01348 ;.
Length = 471
Score = 43.9 bits (102), Expect = 3e-04
Identities = 21/21 (100%), Positives = 21/21 (100%)
Frame = +2
Query: 24 VIKTRLQVAARAGQTTYNGVT 44
VIKTRLQVAARAGQTTYNGVT
Sbjct: 2 VIKTRLQVAARAGQTTYNGVT 64
Shown above are the two ESTs that were at the existing boundaries of the small gap. Look at the first one W76821.1 (this is an accession number for the sequence. It has a version number. Some version numbers may be as high as 29). Notice on the 3rd line that the Length is 455 nucleotides. Now look at the alignment. The upper sequence in the alignment is your query sequence. The lower sequence is the mouse EST hit. The line in between shows the matches between the two sequences. Look at the numbering on the lower sequence from 384 to 452. These are nucleotide numbers, 3/amino acid. 452 is only three nucleotides from the end of the sequence. So this sequence stops where our gap was. The second EST W77458.1 starts where the gap ends. It has only one nucleotide protruding into the gap. If you click on the accession number of the first sequence it takes you to the sequence entry which was created on June 20, 1996. The second est was created on the same date. Notice that both ESTs have another identifier that looks like this: me73f10.r1. The r1 on the end of this name indicates it was sequenced from a reverse primer. This information may be valuable, since the insert of this clone may have been sequenced from the opposite end of the clone. That sequence would have a different accession number, but it would have the same clone name with .s1 at the end instead of .r1. The s1 indicates a standard read from a forward primer. The two sequences would be from the same clone, but from opposite ends. This can unite sequence fragments that do not overlap in the middle. You can search for the clone name (without the .r1 part) in the NCBI homepage search window (select nucleotide in the pull down menu first). If you find the .s1 match then you have more sequence information to use.
We have seen that the gap in the original sequence was caused by these sequences ending just a little short of overlapping. The two best hits in this search do span the gap, allowing the missing sequence to be filled in.
>gi|9904727|gb|BE624311.1|BE624311 uu45a06.y1 Soares_thymus_2NbMT Mus musculus cDNA clone
IMAGE:3374866 5' similar to TR:O14575 O14575 SIMILAR TO
ADP/ATP CARRIER PROTEINS ;.
Length = 480
Score = 80.1 bits (196), Expect = 4e-15
Identities = 44/52 (84%), Positives = 44/52 (84%), Gaps = 8/52 (15%)
Frame = +1
Query: 1 ANEDGQVSPGSLLLAGAIAGMPA--------VIKTRLQVAARAGQTTYNGVT 44
ANEDGQVSPGSLLLAGAIAGMPA VIKTRLQVAARAGQTTYNGVT
Sbjct: 283 ANEDGQVSPGSLLLAGAIAGMPAASLVTPADVIKTRLQVAARAGQTTYNGVT 438
>gi|3519846|gb|AI119522.1|AI119522 uf04h04.y1 Sugano mouse liver mlia Mus musculus cDNA clone
IMAGE:1499671 5' similar to WP:K02F3.2 CE01348 ;.
Length = 670
Score = 80.1 bits (196), Expect = 4e-15
Identities = 44/52 (84%), Positives = 44/52 (84%), Gaps = 8/52 (15%)
Frame = +2
Query: 1 ANEDGQVSPGSLLLAGAIAGMPA--------VIKTRLQVAARAGQTTYNGVT 44
ANEDGQVSPGSLLLAGAIAGMPA VIKTRLQVAARAGQTTYNGVT
Sbjct: 482 ANEDGQVSPGSLLLAGAIAGMPAASLVTPADVIKTRLQVAARAGQTTYNGVT 637
BE624311 uu45a06.y1 was created August 24, 2000. The second one was created Sept. 2, 1998. Note that the clone name has a .y1 after it. That pairs up with a .z1 of the same name. The hit shown below does not come from the same gene. It is nearly identical on the right side, but not on the left. It is from sequence 90 in the alignment (see above). You need to be careful, especially with short sequences, that you are not making chimeric or hybrid sequences when you assemble protein sequences from fragments.
>gi|10753840|gb|BF022507.1|BF022507 uy43g06.y1 NCI_CGAP_Lu30 Mus musculus cDNA clone IMAGE:3662362 5'
similar to TR:O75746 O75746 ARALAR1 PROTEIN. ;.
Length = 450
Score = 56.6 bits (135), Expect = 5e-08
Identities = 32/51 (62%), Positives = 39/51 (75%), Gaps = 8/51 (15%)
Frame = +3
Query: 1 ANEDGQVSPGSLLLAGAIAGMPA--------VIKTRLQVAARAGQTTYNGV 43
A+E+G+V +LL AGA+AG+PA VIKTRLQVAARAGQTTY+GV
Sbjct: 294 ADENGRVGGINLLTAGALAGVPAASLVTPADVIKTRLQVAARAGQTTYSGV 446
That was an easy example of using the EST database to find a missing sequence fragment from a partial gene. Now we will try a harder problem with a larger gap, also in a mouse carrier. Go back to the sequence alignment link at the top of this page and search for -SFLAG in the alignment. You should find sequence 96 as shown below.
segment 3
94A ----------PRKSDGSGEAVFYWSLIAGLLSGMTSAFMVTPFDVVKTRLQADGEKK--------FKGIM 0
95 ------------FNELAGKASFAHSFVSGCVAGSIAAVAVTPLDVLKTRIQTLKKGLGE----DMYSGIT 0
96 ------------------------SFLAGCVAGSAAAVAVNPCDVVKTRLQSLERGVNE----DTYSGFL 0
96A ----------PRRNDGSGEAVFWCSFLAGLAAGSTAALAVNPFDVVKTRLQAIKKADGE----KEFKGIS 0
This gap extends upstream over several pages of the alignment. For your convenience they are shown here with some surrounding sequence. This will be important later. Notice that sequence 95 and 96 in the first segment below are nearly 100% identical. Sequence 95 is a human sequence and it is the ortholog of the mouse sequence with the gap. That will be useful to us.
segment 1
94 GMYRGAAVNLTLVTPEKAIKLAANDFFRHQLS-KDG-----QKLTLLKEMLAGCGAGTCQVIVTTPMEML 112
94A GMYRGSAVNIVLITPEKAIKLTANDFFRYHLASDDG------VIPLSRATLAGGLAGLFQIVVTTPMELL 0
95 GMYRGAAVNLTLVTPEKAIKLAANDFFRRLLM-EDG-----MQRNLKMEMLAGCGAGMCQVVVTCPMEML 0
96 GMYRGAAVNLTLVTPEKAIKLAANDFLRH----------------------------------------- 0
96A GMYRGSGVNILLITPEKAIKLTANDYFRHKLTTKDG------KLPLTSQMVAGGLAGAFQIIVTTPMELL 0
segment 2
94 KIQLQDAGRIAAQRKILAPRSTATQLTRDLLRSRG-IAGLYKGLGATLLRDVPFSVVYFPLFANLNQLG- 0
94A KIQMQDAGRVDRAAGREVKTITALGLTKTLLRERG-IFGLYKGVGATGVRDITFSMVYFPLMAWINDQG- 0
95 KIQLQDAGRLAVHHQGSARRPSATLIAWELLRTQG-LAGLYRGLGATLLRDIPFSIIYFPLFANLNNLG- 0
96 ---------------------------------------------------------------------- 0
96A KIQMQDAGRVAKLAGKTVEKVSATQLASQLIKDKG-IFGLYKGIGATGLRDVTFSIIYFPLFATLNDLG- 0
Try to do the same technique as before. Copy the beginning and end of sequence 96 and place them in the tblastn window. Select mouse ESTs and turn off the filter. Do the blast. The results will show hits that extend into the gap region.
>gi|11620519|gb|BF533156.1|BF533156 602073695F1 NCI_CGAP_Li9 Mus musculus cDNA clone IMAGE:4210775 5'.
Length = 890
Score = 86.3 bits (212), Expect = 4e-17
Identities = 42/42 (100%), Positives = 42/42 (100%)
Frame = +1
Query: 30 SFLAGCVAGSAAAVAVNPCDVVKTRLQSLERGVNEDTYSGFL 71
SFLAGCVAGSAAAVAVNPCDVVKTRLQSLERGVNEDTYSGFL
Sbjct: 124 SFLAGCVAGSAAAVAVNPCDVVKTRLQSLERGVNEDTYSGFL 249
>gi|10379135|gb|BE861312.1|BE861312 UI-M-AK0-adi-c-11-0-UI.r1 NIH_BMAP_MHY Mus musculus cDNA clone
UI-M-AK0-adi-c-11-0-UI 5'.
Length = 538
Score = 65.5 bits (158), Expect = 8e-11
Identities = 39/73 (53%), Positives = 43/73 (58%), Gaps = 13/73 (17%)
Frame = +3
Query: 1 GMYRGAAVNLTLVTPEKAIKLAANDFLRH-------------SFLAGCVAGSAAAVAVNP 47
GMYRGAAVNLTLVTPEKAIKLAANDFLR LAGC AG P
Sbjct: 309 GMYRGAAVNLTLVTPEKAIKLAANDFLRQLLMQDGTQRNLKMEMLAGCGAGICQVGITCP 488
Query: 48 CDVVKTRLQSLER 60
+++K +LQ R
Sbjct: 489 KEMLKIQLQDAGR 527
The first hit above (BF533156) extends 123 bp into the gap upstream. You can see this from the numbering of the lower sequence starting at 124. To get this upstream sequence you must click on the accession number from the blast search and go to the sequence entry. Copy the sequence and take it to the EXPASY DNA translator
Paste it in the window and then translate it. Compare the translations to the mouse and the human sequence above it to find the correct frame.
The DNA sequence of BF533156
GCCAACCTGAATCAGCTGGGCAAACAGGGGGAAGTAGACAATGGAGAAGGGAACATCCCC
CTGTTTGCCAACCTGAATCAGCTGGGCCGCCCATCCTCTGAGGAGAAGTCGCCTTTCTAT
GTGTCCTTCCTAGCAGGCTGCGTGGCTGGGAGTGCAGCCGCTGTGGCCGTCAACCCTTGT
GATGTGGTGAAGACTCGGCTCCAGTCCCTTGAGAGAGGTGTTAATGAGGACACTTACTCT
GGGTTTCTGGACTGTGCAAGGAAGATCTGGAGACATGAAGGTCCCTCAGCCTTCCTGAAA
GGCGCATACTGCCGTGCGCTGGTCATTGCCCCGCTGTTTGGCATCGCCCAGGTGGTCTAC
TTTCTGGGCATTGCCGAGTCCCTGCTGGGGCTGCTGCAAGAACCCCAGGCCTGAGCCCAT
GGCTGCTTCTCTCCAGCCTATGGGCAGGGGCCAGAACAGGGTGACCAGCACAAGCCTGAG
GAGGAGTGGTCTCTCCCCGGTCCTCCTCATTAAGATGGGAAGGCAAGGGGAGGGTGCAGG
GTCCACATGGGTGATGCACACATAAGCCCCTGTGTGGTCCTGAAGGGACAACAAATGGGA
TCGAGGTCTTATCTATGTAGAAAATGCAGAAATCTGTACATCCCTCAAGCCAGTTCTGTC
CCATCCTTGTTACTCAAACCCAGTCCACTGGCTGAACACCCATGGGACAGAGCTGGTCTC
TGGGTGGGGGCCCCAGGCCTGGTTTGGGAGGGGGACCTACCTGGGGTTCACTGGGCCTGG
CCCTGGGGGCCCTGGCTTCCATAGGGGCCCACCCCCGATTTTTTGGGTTCCCCGCCAGGG
GTCTCGGCCGGCGAGCTTGCCGGTGGTCCCCTCGACCTGTCCCCGCTTGG
Only the first three lines are needed to include the first 124 bp.
Translation of the first 180bp phase 1 has the known sequence SFLAG...
And no stop codons, so this is probably correct if there are no frameshifts.
Compare it to the human sequence.
Phase 1
ANLNQLGKQGEVDNGEGNIPLFANLNQLGRPSSEEKSPFYVSFLAGCVAGSAAAVAVNPC
Phase 2
PT*ISWANRGK*TMEKGTSPCLPT*ISWAAHPLRRSRLSMCPS*QAAWLGVQPLWPSTL
Phase 3
QPESAGQTGGSRQWRREHPPVCQPESAGPPIL*GEVAFLCVLPSRLRGWECSRCGRQPL
Phase 1 mouse
ANLNQLGKQGEVDNGEGNIPLFANLNQLGRPSSEEKSPFYVSFLAGCVAGSAAAVAVNPC
||||||| || | | || |||||| ||||| |
YRGLGATLLRDIPFSIIYFPLFANLNNLGFNELAGKASFAHSFVSGCVAGSIAAVAVTPL
Human 95
At least from PLFAN the sequences match, but upstream they do not and this region must be considered suspect. You have now filled in part of the gap. The region between FANLNNLG and GCVAG is not very strong and may contain a frameshift. Watch for better matches in this region.
Look at the other hit BE861312. This has sequence going into the gap from the other side. Compare the sequence with the human sequence. The top sequence is from the blast output starting after the known sequence NDFLR
mouse
QLLMQDGTQRNLKMEMLAGCGAGICQVGITCPKEMLKIQLQDAGR
||| || ||||||||||||||| ||| ||||||||||||||||
RLLMEDGMQRNLKMEMLAGCGAGMCQVVVTCPMEMLKIQLQDAGR
Human 95
They are almost identical and so the frame is correct. This fills in much of the gap from the other side. There are only about 48 amino acids missing in the middle now. To try to fill this gap you can repeat the search of the EST database or you can go to another section to find the missing part, the HTGS section. I recommend this. You will find genomic DNA with introns here, so you need to have the human protein sequence to help identify the missing mouse exon sequence. Construct a hybrid sequence with the new mouse sequence you just found and human from seq 95 to fill in the gap. Then blast the mouse HTGS section of Genbank. In the sequence below human is in lower case.
RLLMEDGMQRNLKMEMLAGCGAGMCQVVVTCPMEMLKIQLQDAGR
lavhhqgsarrpsatliawellrtqglaglyrglgatllrdipfsiiyf
PLFANLNNLGFNELAGKASFAHSFVSGCVAGSIAAVAVTPL
The result from this search shows a good match which is probably to the same gene. There are some differences, however this is a genomic sequence and the gene is on a 90,000 bp contig that is probably more accurate than the EST sequences. There are also three different exons here (see the nucleotide numbering). One of the regions with the greatest number of sequence differences is the region that looked like it might contain frameshifts earlier. The differences on the 2nd exon are mouse human differences, because we searched with human sequence, not mouse.
>gb|AC084273.11|AC084273 Mus musculus clone rp23-313e8, WORKING DRAFT SEQUENCE, 5 unordered pieces
Length = 182050
Score = 81.6 bits (200), Expect = 2e-15
Identities = 38/45 (84%), Positives = 40/45 (88%)
Frame = -2
Query: 9 QRNLKMEMLAGCGAGMCQVVVTCPMEMLKIQLQDAGRLAVHHQGS 53
QRNLKMEMLAGCGAG+CQVV+TCPMEMLKIQLQDAGRL H S
Sbjct: 142635 QRNLKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRLGEAHPNS 142501
Score = 58.5 bits (140), Expect = 2e-08
Identities = 30/34 (88%), Positives = 31/34 (90%)
Frame = -3
Query: 55 RRPSATLIAWELLRTQGLAGLYRGLGATLLRDIP 88
RRPSATLIA ELLRTQGL+GLYRGLGATLLR P
Sbjct: 139970 RRPSATLIARELLRTQGLSGLYRGLGATLLR*AP 139869
Score = 92.4 bits (228), Expect = 1e-18
Identities = 44/53 (83%), Positives = 47/53 (88%)
Frame = -3
Query: 83 LLRDIPFSIIYFPLFANLNNLGFNELAGKASFAHSFVSGCVAGSIAAVAVTPL 135
L RDIPFSIIYFPLFANLN LG +EL GKASF HSFV+GC AGS+AAVAVTPL
Sbjct: 139628 LYRDIPFSIIYFPLFANLNQLGVSELTGKASFTHSFVAGCTAGSVAAVAVTPL 139470
Probable sequence after assembly
QRNLKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRLXXXXXXX
RRPSATLIARELLRTQGLSGLYRGLGATLLRDIPFSIIYFPLFANLNQLGVSELTGKASFTHSFVAGCTAGSVAAVAVTPL
The sequence is almost done now, but there is the small matter of an intron exon joint to be settled. Introns are intervening sequences in genes that separate exons of coding sequence. Introns ususally begin with GT and end with AG. The human sequence has a phase 1 intron after DAGRL and the mouse is expected to have the same intron location. However, the sequence between the human and mouse does not match here for 7 amino acids. It would be desirable to find an EST for mouse that bridged this gap. The following search shows that there is an EST that does this and this sequence indicates that the mouse sequence is longer than the human sequence from the alignment.
>gb|AI848503.1|AI848503 UI-M-AP1-agf-c-11-0-UI.s2 NIH_BMAP_MST_N Mus musculus cDNA clone
UI-M-AP1-agf-c-11-0-UI 3'.
Length = 438
Score = 200 bits (508), Expect = 5e-51
Identities = 106/131 (80%), Positives = 106/131 (80%), Gaps = 18/131 (13%)
Frame = -3
Query: 1 QRNLKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRLXX------------------XX 42
QRNLKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRL
Sbjct: 415 QRNLKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRLAVCHQASASATPTSRPYSTGST 236
Query: 43 XXXRRPSATLIARELLRTQGLSGLYRGLGATLLRDIPFSIIYFPLFANLNQLGVSELTGK 102
RRPSATLIARELLRTQGLSGLYRGLGATLLRDIPFSIIYFPLFANLNQLGVSELTGK
Sbjct: 235 STHRRPSATLIARELLRTQGLSGLYRGLGATLLRDIPFSIIYFPLFANLNQLGVSELTGK 56
Query: 103 ASFTHSFVAGC 113
ASFTHSFVAGC
Sbjct: 55 ASFTHSFVAGC 23
This actually identifies a flaw in the alignment where a part of the human gene was left out because it was too long to fit into the existing alignment. The match below is human on the bottom and mouse on the top, showing the extra sequence also exists in a human EST.
>gb|BF329616.1|BF329616 CM2-BN0273-080600-227-b08 BN0273 Homo sapiens cDNA.
Length = 298
Score = 167 bits (423), Expect = 9e-41
Identities = 84/95 (88%), Positives = 88/95 (92%)
Frame = -3
Query: 4 LKMEMLAGCGAGICQVVITCPMEMLKIQLQDAGRLAVCHQASASATPTSRPYSTGSTSTH 63
LKMEMLAGCGAG+CQVV+TCPMEMLKIQLQDAGRLAV HQ SASA TSR Y+TGS STH
Sbjct: 287 LKMEMLAGCGAGMCQVVVTCPMEMLKIQLQDAGRLAVHHQGSASAPSTSRSYTTGSASTH 108
Query: 64 RRPSATLIARELLRTQGLSGLYRGLGATLLRDIPF 98
RRPSATLIA ELLRTQGL+GLYRGLGATLLRDIPF
Sbjct: 107 RRPSATLIAWELLRTQGLAGLYRGLGATLLRDIPF 3
This effort now successfully fills in the gap in mouse sequence 96.
Assignment 1
To test out your new skills at blast searching lets assemble a gene or two from ESTs. Since there are about 10 of us registered for the course I have assigned each of you two Arabidopsis (or rice) sequences to search against the Zea mays EST database to find the maize orthologs of these sequences. The whole Arabidopsis sequences can be obtained at another linked file which has the FASTA format of the selected rice P450 sequences.
E. Akano 75A11 77A9
C. Bricker 77B2 79A7
S. Chintalapudi 89B1 96B2
G. Kuntamallappanavar 89C1 96E1
B. Manda 97B4 701A6
A. Pandey 97C2 703A3
P. Sharma 706C1 707A5
Guest 1 710A5
Guest 2 728B1
Guest 3 735A3
If you finish and would like more to do you can try the same sequences
searched against the wheat ESTs Triticum aestivum. These should be available since
there are over 1 million wheat ESTs.
Go to the FASTA file and search for your sequence name. The CYP prefix is part of the name for Cytochrome P450, but you don't need to search for the whole name. Copy the sequence and take it to the NCBI blast page. Do a tblastn search of the est others database division (not just EST, since that will include mouse and human) and set the species in the organism window to Zea mays. When you get your results, scroll down and look for those sequences with the best match. Shorter sequences will appear later even if they are better matches than longer sequences, so don't look just at the top. When you find your top two or three best matches, you can check if they are the orthologs of the rice sequence by blast searching against the rice P450 data set. I have a server just for this puprose at P450 Blast server. Paste your sequence in this window, select rice from the pull down menu and run the blast. The default database in this blast server is all the human P450 sequences, so you will need to change from the default. If your best hit to rice P450s is the sequence you started with then you probably have the ortholog. This is called the best reciprocal blast hit. Some sequences may not have ortholog hits in the rice ESTs. Do not despair. Just find the matches that are the best you can find to your starting sequence.
Another search option in the P450 blast page is to use the program blastx with the full length EST nucleotide sequence. You can get the nucleotide sequence by clicking on the hyperlinked accession number in the blast output from your NCBI search. You can compare it to rice on the same blast server. Your Zea mays sequences should match best to the rice P450 sequences.
Important tip: When working with sequences in a word processor set the font to
Courier 9 point. This is a fixed space font that will keep your alignments properly aligned. The 9 point size prevents wrapping of blast results when you paste them into a document.
This is an example of the output from a human CYP20 search of Salmo salar ESTs.
The starting sequence was human CYP20. You can check that by comparing the top (query sequence) with the P450 blast server. Genes are genes, so for this example I used human and fish. The results will be very similar when you use rice and Zea mays.

>gi|117525684|gb|EG857411.1| UniGene info EST_ssal_eve_50960 ssaleve thyroid Salmo salar cDNA Salmo salar
cDNA clone ssal_eve_569_158_fwd 5', mRNA sequence.
Length=735
Score = 266 bits (679), Expect = 1e-70, Method: Compositional matrix adjust.
Identities = 148/222 (66%), Positives = 180/222 (81%), Gaps = 1/222 (0%)
Frame = +2
this is the 5th hit on the left side of the graphic (N-terminal part of CYP20)
Query 1 MLDFAIFAVTFllalvgavlylyPASRQAAGIPGITPTEEKDGNLPDIVNSGSLHEFLVN 60
MLDFAIFAVTF++ LVGAVLYLYP+SR A+GIPG+ PTEEKDGNL DIVN GSLHEFL +
Sbjct 35 MLDFAIFAVTFVIFLVGAVLYLYPSSRSASGIPGLNPTEEKDGNLQDIVNRGSLHEFLAS 214
Query 61 LHERYGPVVSFWFGRRLVVSLGTVDVLKQHINPNKTSDPFETMLKSLLRYQSG-GGSVSE 119
LH ++GPV SFWFG R VVSLG+VD L+QHINPN+T+D FETMLKSLL YQSG GG +E
Sbjct 215 LHGQFGPVASFWFGGRPVVSLGSVDQLRQHINPNRTTDSFETMLKSLLGYQSGTGGGATE 394
Query 120 NHMRKKLYENGVTDSLKSNFAlllklseelldkwlsYPETQHVPLSQHMLGFAMKSVTQM 179
MRKKLYE+ V ++ + NF +LLKL EEL+ KW S+P+ QH PL H+ G AMK+VTQ+
Sbjct 395 AVMRKKLYESAVNNTPEKNFPMLLKLVEELVGKWQSFPKDQHTPLCAHLQGLAMKAVTQL 574
Query 180 VMGSTFEDDQEVIRFQKNHGTVWSEIGKGFLDGSLDKNMTRK 221
+G F +D EVI F+KNH +WSEIGKG+LDGS++K+ RK
Sbjct 575 ALGDRFRNDAEVIGFRKNHEAIWSEIGKGYLDGSMEKSSIRK 700
>gi|89852388|gb|DY708511.1| UniGene info EST_ssal_rgb2_64250 ssalrgb2 mixed_tissue Salmo salar cDNA Salmo salar cDNA clone ssal_rgb2_603_269_rev 5', mRNA sequence. Length=866 Score = 355 bits (911), Expect = 2e-97, Method: Compositional matrix adjust. Identities = 175/287 (60%), Positives = 230/287 (80%), Gaps = 2/287 (0%) Frame = +3
This is the top hit in the graphic (middle part of CYP20)
Query 31 GIPGITPTEEKDGNLPDIVNSGSLHEFLVNLHERYGPVVSFWFGRRLVVSLGTVDVLKQH 90
GIPG+ PTEEKDGNL DIVN GSLHEFL +LH ++GPV SFWFGR VVSLG+VD L+QH
Sbjct 3 GIPGLNPTEEKDGNLQDIVNRGSLHEFLASLHGQFGPVASFWFGGRPVVSLGSVDQLRQH 182
Query 91 INPNKTSDPFETMLKSLLRYQSG-GGSVSENHMRKKLYENGVTDSLKSNFAlllklseel 149
INPN+T+D FETMLKSLL YQSG-GG +E MRKKLYE+ V ++L+ NF +LLKL EEL
Sbjct 183 INPNRTTDSFETMLKSLLGYQSGTGGGATEAVMRKKLYESAVNNTLEKNFPMLLKLVEEL 362
Query 150 ldkwlsYPETQHVPLSQHMLGFAMKSVTQMVMGSTFEDDQEVIRFQKNHGTVWSEIGKGF 209
+ KW S+P+ QH PL H+LG AMK+VTQ+ +G F +D EVI F+KNH + WSEIGKG+
Sbjct 363 VGKWQSFPKDQHTPLCAHLLGLAMKAVTQLALGDRFRNDAEVIGFRKNHEAIWSEIGKGY 542
Query 210 LDGSLDKNMTRKKQYEDALMQLESVLRNIIKERKGRNFSQHIFIDSLVQGNLNDQQILED 269
LDGS++K+ RK+ YE AL ++E+VL ++K+RKG+ SQ F+D+L+Q NL ++Q++ED
Sbjct 543 LDGSMEKSSIRKEHYESALAEMETVLMSVAKDRKGQR-SQTAFVDTLLQSNLTERQVMED 719
Query 270 SMIFSLASCIITAKLCTWAICFLTTSEEVQKKLYEEINQVFGNGPVT 316
SM+F+LA C+ITA LC WA+ FL+TSE+VQ+KL++E+ V G+ PV+
Sbjct 720 SMVFTLAGCVITANLCIWAVHFLSTSEDVQEKLHQELEDVLGSEPVS 860
>gi|24393371|gb|CA063128.1| UniGene infoGeo ssalrgb509318 mixed_tissue Salmo salar cDNA, mRNA sequence.
Length=695
Score = 201 bits (512), Expect = 3e-51, Method: Compositional matrix adjust.
Identities = 103/168 (61%), Positives = 136/168 (80%), Gaps = 0/168 (0%)
Frame = -1
This is the 5th hit on the right side of the graphic (C-terminal part of CYP20)
Query 294 TSEEVQKKLYEEINQVFGNGPVTPEKIEQLRYCQHVLCETVRTAKLTPVSAQLQDIEGKI 353
TSE+VQ+KL++E+ V G+ PV+ +KI QLRY Q VL ETVRTAKLTP++A+LQ EGK+
Sbjct 695 TSEDVQEKLHQELEDVLGSEPVSLDKIPQLRYFQQVLNETVRTAKLTPIAARLQXNEGKV 516
Query 354 DRFIIPRETLVLYALGVVLQDPNTWPSPHKFDPDRFDDELVMKTFSSLGFSGTQECPELR 413
D+ IIP+ETLV+YALGVVLQD +TW P+KFDPDRF ++ K+FS LGFSG Q CPELR
Sbjct 515 DQHIIPKETLVIYALGVVLQDADTWSCPYKFDPDRFTEDSARKSFSLLGFSGNQACPELR 336
Query 414 FAYMVTTvllsvlvkrlhllsvEGQVIETKYELVTSSREEAWITVSKR 461
FAY V TV+LS +V++L L V+GQV+E + ELV++ +++ WITVS+R
Sbjct 335 FAYTVATVVLSTVVRQLKLYQVKGQVVEARSELVSTPKDDTWITVSRR 192
Assembled salmon CYP20 sequence from the three ESTs (bottom lines)
Overlaps have been removed
MLDFAIFAVTFVIFLVGAVLYLYPSSRSASGIPGLNPTEEKDGNLQDIVNRGSLHEFLAS
LHGQFGPVASFWFGGRPVVSLGSVDQLRQHINPNRTTDSFETMLKSLLGYQSGTGGGATE
AVMRKKLYESAVNNTPEKNFPMLLKLVEELVGKWQSFPKDQHTPLCAHLQGLAMKAVTQL
ALGDRFRNDAEVIGFRKNHEAIWSEIGKGYLDGSMEKSSIRK
EHYESALAEMETVLMSVAKDRKGQRSQTAFVDTLLQSNLTERQVMED
SMVFTLAGCVITANLCIWAVHFLSTSEDVQEKLHQELEDVLGSEPVS
LDKIPQLRYFQQVLNETVRTAKLTPIAARLQXNEGKV
DQHIIPKETLVIYALGVVLQDADTWSCPYKFDPDRFTEDSARKSFSLLGFSGNQACPELR
FAYTVATVVLSTVVRQLKLYQVKGQVVEARSELVSTPKDDTWITVSRRS*
Note: I added the last amino acid and the stop codon from the translation of the CA063128 DNA sequence.
Use the bottom protein sequence (the sequence marked Sbjct) to search with the human P450 blast server. Or you can click on the accession number of your NCBI blast hit to get the nucleotide sequence. The whole nucleotide sequence can be pasted in the P450 Blast server window and the program must be set to blastx instead of blastp. Searches of Human or another vetebrate like Fugu (in the pull down menu) will align the translated sequence against the P450 database entries.
The output from this search will look something like this
Whole salmon sequence compared to human in the P450 blast server
Note: the top seq is now the salmon sequence.
>CYP20 AC011737.8 chr 2 (missing exons 12,13) AC080075.2 (missing exons
1,7,8)
Length = 462
Score = 595 bits (1534), Expect = e-173
Identities = 288/462 (62%), Positives = 373/462 (80%), Gaps = 2/462 (0%)
Query: 1 MLDFAIFAVTFVIFLVGAVLYLYPSSRSASGIPGLNPTEEKDGNLQDIVNRGSLHEFLAS 60
MLDFAIFAVTF++ LVGAVLYLYP+SR A+GIPG+ PTEEKDGNL DIVN GSLHEFL +
Sbjct: 1 MLDFAIFAVTFLLALVGAVLYLYPASRQAAGIPGITPTEEKDGNLPDIVNSGSLHEFLVN 60
Query: 61 LHGQFGPVASFWFGGRPVVSLGSVDQLRQHINPNRTTDSFETMLKSLLGYQSGTGGGATE 120
LH ++GPV SFWFG R VVSLG+VD L+QHINPN+T+D FETMLKSLL YQSG GG +E
Sbjct: 61 LHERYGPVVSFWFGRRLVVSLGTVDVLKQHINPNKTSDPFETMLKSLLRYQSG-GGSVSE 119
Query: 121 AVMRKKLYESAVNNTPEKNFPMLLKLVEELVGKWQSFPKDQHTPLCAHLQGLAMKAVTQL 180
MRKKLYE+ V ++ + NF +LLKL EEL+ KW S+P+ QH PL H+ G AMK+VTQ+
Sbjct: 120 NHMRKKLYENGVTDSLKSNFALLLKLSEELLDKWLSYPETQHVPLSQHMLGFAMKSVTQM 179
Query: 181 ALGDRFRNDAEVIGFRKNHEAIWSEIGKGYLDGSMEKSSIRKEHYESALAEMETVLMSVA 240
+G F +D EVI F+KNH +WSEIGKG+LDGS++K+ RK+ YE AL ++E+VL ++
Sbjct: 180 VMGSTFEDDQEVIRFQKNHGTVWSEIGKGFLDGSLDKNMTRKKQYEDALMQLESVLRNII 239
Query: 241 KDRKGQR-SQTAFVDTLLQSNLTERQVMEDSMVFTLAGCVITANLCIWAVHFLSTSEDVQ 299
K+RKG+ SQ F+D+L+Q NL ++Q++EDSM+F+LA C+ITA LC WA+ FL+TSE+VQ
Sbjct: 240 KERKGRNFSQHIFIDSLVQGNLNDQQILEDSMIFSLASCIITAKLCTWAICFLTTSEEVQ 299
Query: 300 EKLHQELEDVLGSEPVSLDKIPQLRYFQQVLNETVRTAKLTPIAARLQXNEGKVDQHIIP 359
+KL++E+ V G+ PV+ +KI QLRY Q VL ETVRTAKLTP++A+LQ EGK+D+ IIP
Sbjct: 300 KKLYEEINQVFGNGPVTPEKIEQLRYCQHVLCETVRTAKLTPVSAQLQDIEGKIDRFIIP 359
Query: 360 KETLVIYALGVVLQDADTWSCPYKFDPDRFTEDSARKSFSLLGFSGNQACPELRFAYTVA 419
+ETLV+YALGVVLQD +TW P+KFDPDRF ++ K+FS LGFSG Q CPELRFAY V
Sbjct: 360 RETLVLYALGVVLQDPNTWPSPHKFDPDRFDDELVMKTFSSLGFSGTQECPELRFAYMVT 419
Query: 420 TVVLSTVVRQLKLYQVKGQVVEARSELVSTPKDDTWITVSRR 461
TV+LS +V++L L V+GQV+E + ELV++ +++ WITVS+R
Sbjct: 420 TVLLSVLVKRLHLLSVEGQVIETKYELVTSSREEAWITVSKR 461
For the assignment, please turn in the following for each of your two P450 sequences:
1) The accession numbers of your maize hits that you feel are your best hits. The accession number in the example above is EG857411. I cannot use your data if you do not supply the accession number.
2) The sequence name you were using for the search (CYP51G1 or CYP701A3 etc.)
3) The output from the human P450 blast server that shows the best match to your Zea mays EST.
4) your name
This will look like this:
salmon EST sequence EG857411
human starting sequence CYP20
>CYP20 AC011737.8 chr 2 (missing exons 12,13) AC080075.2 (missing exons
1,7,8)
Length = 462
Score = 595 bits (1534), Expect = e-173
Identities = 288/462 (62%), Positives = 373/462 (80%), Gaps = 2/462 (0%)
Query: 1 MLDFAIFAVTFVIFLVGAVLYLYPSSRSASGIPGLNPTEEKDGNLQDIVNRGSLHEFLAS 60
MLDFAIFAVTF++ LVGAVLYLYP+SR A+GIPG+ PTEEKDGNL DIVN GSLHEFL +
Sbjct: 1 MLDFAIFAVTFLLALVGAVLYLYPASRQAAGIPGITPTEEKDGNLPDIVNSGSLHEFLVN 60
Query: 61 LHGQFGPVASFWFGGRPVVSLGSVDQLRQHINPNRTTDSFETMLKSLLGYQSGTGGGATE 120
LH ++GPV SFWFG R VVSLG+VD L+QHINPN+T+D FETMLKSLL YQSG GG +E
Sbjct: 61 LHERYGPVVSFWFGRRLVVSLGTVDVLKQHINPNKTSDPFETMLKSLLRYQSG-GGSVSE 119
Your Name
Do this for each of your best hits (up to three).
Optional: walk along the Zea mays EST by using the translated protein sequence you found as the starting point for another search of the Zea mays EST database. This new search will find overlapping ESTs that may let you walk to the beginnning or end of the Zea P450. Try to assemble a whole P450 from overlapping ESTs. Please keep track of the accession numbers and turn them in with your sequences.
We did this in the example above by using the graphic to pick out overlapping ESTs that would continue the sequence.
Links for this class
NCBI main page
http://www.ncbi.nlm.nih.gov/
NCBI.
TBLASTN page at NCBI
http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?CMD=Web&LAYOUT=TwoWindows&AUTO_FORMAT=Semiauto&ALIGNMENTS=50&ALIGNMENT_VIEW=Pairwise&CLIENT=web&DATABASE=nr&DESCRIPTIONS=100&ENTREZ_QUERY=(none)&EXPECT=10&FILTER=L&FORMAT_OBJECT=Alignment&FORMAT_TYPE=HTML&GENETIC_CODE=0&HITLIST_SIZE=100&NCBI_GI=on&PAGE=Translations&PROGRAM=tblastn&SERVICE=plain&SET_DEFAULTS.x=23&SET_DEFAULTS.y=10&SHOW_OVERVIEW=on&UNGAPPED_ALIGNMENT=no&END_OF_HTTPGET=Yes
TBLASTN
NCBI BLAST page
http://www.ncbi.nlm.nih.gov/BLAST/
NCBI Blast page
P450 Blast server
http://blast.uthsc.edu/
P450 Blast server
EXPASY DNA translator
http://ca.expasy.org/tools/dna.html
EXPASY DNA translator
Mito carrier sequence alignment
http://drnelson.uthsc.edu/car202.class.html
sequence alignment
Rice cytochrome P450 sequences
http://drnelson.uthsc.edu/rice.search.seqs.htm
FASTA format of the selected rice P450 sequences.
my email address
[email protected]