Bioinformatics and mining the human genome
Stresa Italy, July 10, 2000 MDO2000 meeting
David Nelson
Today, genome projects are the fashion. The news is full of the race to finish the human genome. Two weeks ago, on June 26, Francis Collins and Craig Venter stood with President Clinton in the White House as he heralded the completion of a rough draft of the human genome. If one knows where to look, the progress can be monitored on genome meters like this one from the Arabidopsis genome project, (92.9% complete on July 25) slide 1 NCBI has a similar meter for the Human Genome Project slide 2. Here we see that on June 25, the human genome was about 21% finished and about 66% more was in draft form (meaning 99.9% accuracy or better). With all this attention given to these genome projects, don’t be surprised if you get a notice in the mail for a new journal called Nature Genomics.
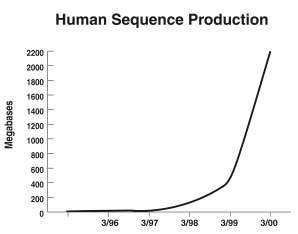
I don’t mean to downplay the importance of all this work. It has been and continues to be an exciting time, one that will not be repeated. Once the human and mouse genomes are done and the genes have been identified by comparison of the two genomes, much of the excitement will pass. But for now, we are in the middle of it. Bets are actually being taken on the number of human genes, and the number ranges from 34000 to 153000. The human gene count is a surprisingly elusive and slippery number. Here is a graph taken from the NHGRI showing sequencing progress. Slide 3 As you can see from this figure, most of the human genome sequence has been acquired in the past 15 months, since March 1999. If you had looked for a human gene in Genbank before 1998 you probably would not have found it. If you looked today the chances are about 90% that you would find it.
{kind=link}
How can it be that we have 87% of the human genome sequence, but we cannot agree on the number of genes within a factor of 4 to 5? This paradox begins to reveal the difficulties of analyzing the DNA sequence. For those of you that have looked at the human genome rough draft sequence, you have probably had some frustrations. For those of you who have not, you may not know what is there. This morning I would like to talk about data mining in the human genome and what king of problems you may run into. I will use examples from the cytochrome P450 genes. We are really faced with numerous problems when looking at the rough draft of the human genome. First, most of the sequence is fragmentary. The 66% that is in draft form is at four levels of completion. Phase 0 sequence is the most preliminary. Slide 4 A large BAC clone, is shotgun sequenced and all the unassembled reads from this sequence set are given one accession number and placed in Genbank in the HTGS (high throughput genomic) section. The fragments are unordered and their correct orientation is unknown. The clone size may be as large as 280000 bp and the number of reads in a single entry may be as high as 360. The read lengths are about 600-750 bp. Because P450 genes are moderately long, about 10000 to 40000 bp, the short reads in phase 0 sequence cannot cover a whole gene. One read may only cover one exon or a part of one exon. If there is only one P450 gene on a whole BAC clone it will be possible to assemble the gene based on comparison to other complete P450s and ESTs, but there are likely to be regions missing in the gaps between some of the reads. You also do not know if there is more than one P450 on a clone. The pieces found may belong to two or more genes.
{kind=link}
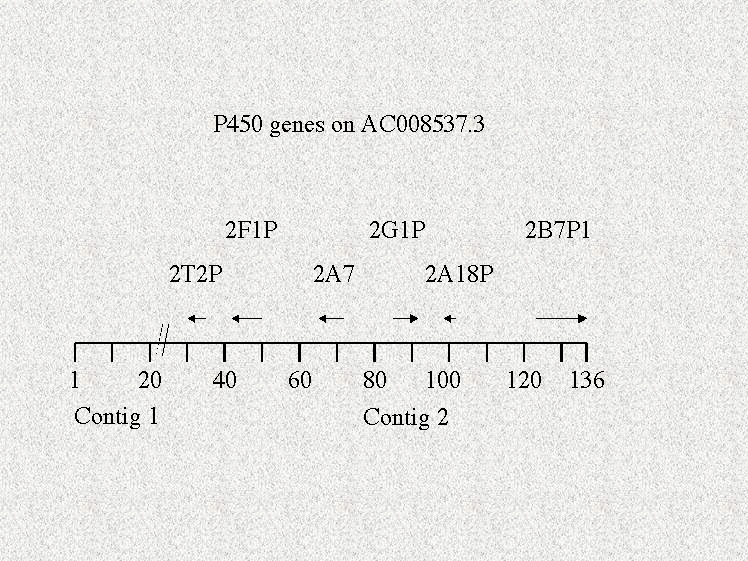
The next level is phase 1. Slide 5 Here the reads have been assembled into contigs by searching for overlaps. The number of contigs drops as the fragments are joined. Phase 1 sequence is still composed of unordered pieces. The accession number is given a version number when it is created. As revisions are made and more fragments are joined the version number is increased. I have seen version numbers reach 28, but they are usually in the 1-6 range. It is still difficult and hazardous to assemble genes from phase 1 sequence. I’ll give you an example from chromosome 19 which has a CYP2 family gene cluster. In late March 2000, this accession number was version 2 and it consisted of 16 unordered pieces. Ten of them had P450 sequence on them, but there was evidence for five different subfamilies being present and probably 7 different genes. At the time it was not possible to assemble all these genes with any confidence. On May 4, the same accession number was at version 3. This was a phase two sequence with only two fragments and they were ordered, so it was now possible to assemble the genes correctly assuming that none of them fell in the single gap or ran off one end or the other. It is also interesting to note that the version 3 sequence was 137000 bp while the earlier version was 170000, so 33000 bp of sequence was discarded. The P450 genes on this sequence look like this now. Slide 6. Six genes are present, but the cluster is not complete. In moving from version 2 to version 3, the 2A6 gene was deleted as well as the C-terminal exon of 2B7P1. The 2A6 gene has reappeared on AC025769.3, but this sequence is labeled as being from chromosome 5. The other sequences seen in this slide are all present on this chromosome 5 sequence, so my guess is that there is a mistake in labeling. Chromosome 19 is probably correct based on other mapping data. The genome sequence is thus in a state of flux.
{kind=link}
{kind=link}
Phase 2 sequence slide 7 has fewer contigs and they are ordered. This is a significant improvement over the unordered pieces of phase 0 and phase 1 sequence. There are still gaps and a long human P450 gene could be missing some exons. The longest human P450 gene I know about so far is CYP5A1. This gene is on a completed genomic sequence [NT_001551] and it spans 197000 bp. If this was in a phase 2 sequence it might be missing half the gene.
{kind=link}
After phase 2 the sequence reaches complete status. There are no more gaps in complete sequences. The last stage in processing the sequence is annotation, with identification of the coding regions, repeats, pseudogenes etc. The annotation is always subject to change. There are often differences of opinion about what constitutes a gene. I have had discussions with genome sequencing experts that say they have found twice as many genes on the completed and annotated human chromosome 22 as the published article claims exist there. In my own experience, I have found two P450 genes in a cluster joined together with the other parts of the two genes overlooked. I have seen this in Drosophila, Arabidopsis and C. elegans, so it is a fairly common problem. This underscores the need for expert human annotation in addition to computer generated annotation.
So far, the problems I have mentioned with assembling P450 genes or any other genes have had to do with the incomplete nature of the sequence data. This will vanish as the sequences are all moved out of phase 0, 1 and 2 to complete status. The problems of gene identification and assembly do not end there. There is still the matter of fate in selection of the DNA to be sequenced. On Chromosome 22, the first completed human chromosome, there is a small cluster of P450 genes. These are the 2D6 gene and two pseudogenes near it. This cluster has been well studied and documented. However, the individual whose DNA was sequenced for the Human Genome Project carried an allele of CYP2D6 called 2D6*5 where the whole 2D6 coding region was deleted and only parts of the pseudogenes remained. So when I tried to find 2D6 on chromosome 22, I could not find it. This is the problem of polymorphisms. I show a comment in this slide that emphasizes the problem. Slide 8 There are numerous deletions. This raises questions about P450s that might be real genes or pseudogenes. On AC008537, there is a gene named CYP2G1P. This gene is normal in every way except it is missing exons 4 and 5. Is this due to a polymorphic deletion in this individual or is CYP2G1P really a pseudogene. We won’t know until the gene can be resequenced from other people. The Celera genomic data is taken from 5 individuals and sequencing on all five was completed June 23, so this question is probably answered in the Celera data set.
{kind=link}
Pseudogenes are also a difficult matter. Currently there are 24 human P450 pseudogenes. These fragments of genes often flank real genes and one has to wonder if there might be alternative splicing in some cases. Among human P450s, there seem to be a large number of 4F and 2C pseudogenes. It is not clear why these parents should throw off so many more pseudogenes than other P450s, but they seem to be more abundant. Pseudogenes may be a problem in humans in general. Another gene that I work on is called the adenine nucleotide translocator or ANT1 and this gene is reported to have at least 9 pseudogenes on various chromosomes. With the draft sequence quality at more than 1 error in 10000bp, it is often difficult to tell a pseudogene from a sequence error. It may take some time or some resequencing to clarify those P450 genes that have one or two in frame stop codons. This is the case for the gene CYP2G2P. Otherwise it looks like a normal gene. Is it real or not?
Another issue that comes up in such a large project is contamination. There is one P450 sequence I found in human draft sequence that did not seem to be human. The best match to this human sequence is a mushroom P450 with 50% sequence identity. Slide 9 I sent notice to Genbank that this was so and asked them to relay the message to the sequencers. When I repeated the search for this sequence in June, I did not find it. Calling up the accession number, I found that the version number had changed and the new sequence was shorter. Slide 10 Looking at the contigs in both the version 2 and version 3 sequence one can see that the first two small contigs have been removed. The P450 sequence was on contig 2. So the sequencers decided to drop those sequences in version 3. I strongly suspect a fungal contamination of the library used in making this clone. Unfortunately, this may affect other sequences in the library that are undetected so far. This is especially hard if the sequence is chimeric, with human DNA on both ends, then it will look like a human sequence and be joined into larger contigs. Errors like this will need to be checked by sequencing multiple individuals to eliminate contaminants.
{kind=link}
{kind=link}
Possibly the most difficult problem in assembling genes from the human genome is not due to sequence errors. Even a perfect sequence would have this problem. It is the difficulty in assembling any gene from genomic sequence, finding the exons, especially the N-terminal exon. One example of this is the new human P450 CYP3A43. This gene is 38000 bp long and it is composed of 13 exons. Slide 11 The first intron is over 8000 bp and the first exon is only 24 amino acids long. Seven exons are less than 40 amino acids long and one is only 18. Luckily the 3A subfamily is pretty well conserved and these short exon fragments can be detected by a systematic search. This would not be the case if the percent identity between a new sequence and some other known sequence was low. In that case one would need to depend on cDNA sequence to help locate the missing pieces, especially the N-terminal. This is exactly the problem with the Dictyostelium discoideum P450 gene sequences. Most of them seem to have a very short and poorly conserved N-terminal exon that just cannot be detected by examining the genomic DNA sequence.
{kind=link}
That brings up the flip side of the human genome project. In addition to sequencing the genomic DNA, nearly two million EST sequences from human cDNA have been deposited in GenBank. These can be very helpful in defining the intron exon boundaries of new genes. More than 1.5 million of these sequences have been incorporated into UNIGENE. Unigene scans the EST sequences and groups them in clusters that are from the same gene, or very similar genes. The May 24 version had 89632 clusters. It is tempting to assume that this means there are about 90000 human genes, but there is not a one to one correspondence between UNIGENE clusters and genes. Over 30000 of these clusters have only one EST sequence and it has been argued recently that many of these are probably accidental missprimings. Serious attempts to estimate the number of human genes ignore these singleton ESTs. That leaves almost 60000 clusters with two or more ESTs. However, different clusters may be from different parts of the same gene.
I have tried to catalog the UNIGENE entries for human, mouse and rat, though this is like shooting at a moving target. Slides 12, 13 see TABLE The data changes so quickly that many of the UNIGENE numbers are retired due to splitting and mergers. Of course pseudogenes that do not make mRNA will not have an entry in UNIGENE. However, there are pseudogenes that do have ESTs such as the new CYP2T2P with 12 ESTs mostly from breast and placenta. This makes one wonder if it is really a pseudogene. A similar gene CYP2T1 has been found in the rat and it is not a pseudogene. So perhaps humans have just recently lost a functional version of this gene. Several other P450 pseudogenes have a UNIGENE entry. These include CYP2B7P1, 2D8P and 3A5P2.
There are also six human P450s that are not pseudogenes yet they have no ESTs associated with them. These are CYP2A13, 2C19, 2F1, 4F22, 7A1 and 27C1. Slide 14 This shows that the EST database does not have every gene represented even though there are 2 million sequences. These 6 P450s represent 11% of the known human P450s, so gene coverage in the human EST database is about 89% based on P450 genes. Searching the human ESTs for a P450 will not always find a good hit, even when there are ESTs for that gene. Some P450s have long 3 prime untranslated sequences and ESTs are often from this part of the gene. CYP26B1 has a 3000bp untranslated 3 prime sequence and no ESTs in the coding region.
{kind=link}
I think I have given you a summary of the problems encountered in data mining the human genome sequence data. I do not want to leave you with the impression that it is not worth the trouble. On the contrary, in the past year, I have found several new P450 genes from the incomplete and sometimes fragmentary human genomic sequences. slide 15 These include, CYP3A43, CYP2G1P, 2G2P, 2T2P, 2T3P, 2U1, 4F22, 4V2 (related to a trout P450 fragment called 4V1), 26B1, and 27C1. This last gene is missing some sequence at the end and in the middle, but it may be an important new member of the mitochondrial P450s.
{kind=link}
The discovery of new human P450s is winding down. With almost 90% of the genome in the databases, one might expect only a 10% increase in gene counts. This is actually misleading, because there are some P450s that are known from cloning that have not been found yet in the genomic sequencing. Slide 16 These make up a part of the missing 10%. CYP2C9, 3A4, 11B1, 11B2 and 26A1 still need to be found in the genomic DNA. (Note, on July 7 CYP3A4 was on AC069294.3. This includes parts of 3A5, 3A7 and 3A43 see file ) We have 53 human P450s now. If we subtract the five given above that are not yet found in the genomic DNA that gives 48 and a 10% increase would be 53 again. Based on these data, I don’t expect to find more than one or two more P450 genes in humans, and those are probably going to be in existing families and subfamilies. There should not be any major surprises in store. Of course, I like surprises. Slide 17
{kind=link}
Note: 2C9 has been found on AL133513 joined to 2C19 to make a hybrid sequence. The 2C cluster has four 2C genes close together. I did a blast of the intron region at the joint of 2C9 and 2C19 and found that most of the intron region about 800bp of the 1160bp region was a line1 repeat element. It had 91% identity to many other line1 repeats in the human genomic sequence. If all four 2C genes have this Line1 repeat in this intron, they could recombine to make six different chimeric P450s that would be functional genes. The original 2C17X gene could have been caused by this phenomenon. The person chosen for sequencing may have a deletion polymorphism of the 3 prime end of 2C9 and the 5 prime end of 2C19. This would be similar to the deletion of 2D6 on chromosome 22.