David Nelson Jan. 21, 2016
This module deals with multiple sequence alignments, how to make them and how to edit them. So far in the course, we have used sequences on my P450 website to find other related sequences. You have learned how to use BLAST searching to find orthologs in ESTs and genomic sequences. The next step in analyzing these new sequences is to incorporate them into an alignment and calculate phylogenetic trees to see the relationships between the sequences. Making a multiple sequence alignment can be done by hand, but it is a slow tedious process. It can also be done by programs, but the results may need some adjusting, since the programs are not perfect.
Before we talk about multiple sequence alignments it is necessary to cover a little history of how optimal alignments of two sequences are made. There are two different approaches to making sequence alignments. One method maximizes the similarity between two sequences. This is done by the Needleman-Wunch algorithm (1970). A different approach minimizes the differences between two sequences. This done by the Sellers algorithm (1974) and the Smith-Waterman algorithm (1981). The Needleman-Wunch algorithm and the Sellers algorithm look very different in how they proceed, but the Sellers algorithm and the Needleman-Wunch algorithm have been proven mathematically to be equivalent (Smith et al. 1981). [For a tutorial on the statistics of sequence comparison see the NCBI link A tutorial on sequence comparison statistical calculations. For a comparison of the methods and their relationship to FASTA and BLAST see these class notes .
The Needleman-Wunch algorithm creates an array with the two sequences on the edges of the array. Each cell in the array represents the comparison of two amino acids, one from each protein. These are given a score based on a similarity matrix like the PAM or BLOSUM matrices. In the example pictured below the scoring matrix uses 1 for a match and 0 for a mismatch. This is called an identity matrix. The PAM and BLOSUM matrices give numerical scores for each amino acid pair. These scores are based on examination of real sequence alignments and statistics on how often one amino acid replaces another in evolution. All the cells in the array are filled following some simple rules and then the path through the array that has the highest scores is identified.
example showing how the program works

This represents the optimum alignment of the two sequences. This operation is dependent on N x M operations where N and M are the lengths of the two sequences. You can see that this will be memory intensive for average sequences of say 500 amino acids, an array of 250,000 elements is needed. The algorithm can be done in n dimensions where n is the number of sequences, but the array sizes keeps growing as a multiple of the sequence lengths. In reality even three 500 amino acid sequences becomes impossible (array size 125 million).
For a nice presentation of the Algorithm see Aoife McLysaght's page from the University of Dublin
Because these rigorous methods cannot be extended to more than two sequences, multiple sequence alignment was done by hand up until about 1987. At that point several people began to ask how can we simplify this problem so it is not so big? The answer is called progressive alignment. In progressive alignment, all the sequences are compared against one another to get a score of relatedness. The matrix of these scores (usually the difference between sequences) is called a difference matrix and this is used to make a tree called a guide tree. In the guide tree the most similar sequences are clustered together. This guide tree is the basis for the multiple sequence alignment. The most similar sequences are aligned one at a time to each other. As sets of aligned sequences are built up, the most similar sets are aligned with each other, until the last sequence is added. At each step, the computational cost is like alignment of only two sequences rather than N sequences. It is possible to do this with large numbers of sequences even on a desktop computer.
The result is not a perfect alignment. The errors that occur early will be propogated throughout the alignment as more sequences are added. To keep the computational cost low, once a gap is added, it cannot be removed. You may imagine that large errors are possible if the two sequences being compared are not of similar length. The program will have to add gaps and the placement of the gaps can be critical for later steps in the alignment process.
Clustal Omega
The most popular program for making multiple sequence alignments is Clustal Omega. This is a newer version of Clustal W and before that ClustalV. There is also an X-windows version called ClustalX and this can be downloaded and installed on a desktop computer from this ClustalX Link. A tutorial on the ClustalX software is available from those wonderful folks at Molecular Systematics and Evolution of Microorganisms. The Natural History Museum, London and Instituto Oswaldo Cruz, FIOCRUZ Rio de Janeiro, Brazil. Click hereFor a cool 9 lecture course in molecular systematics see this link.
Clustal Omega and Clustal W are available at several servers at different web locations. Some of these limit the number of characters you can submit to be aligned and these are not very useful. We have one ClustalW server on a LINUX server and that can be used for the course. Muscle is another alignment program that is very popular and it is considered a bit better than CLUSTAL
ClustalW server in Wageningen UR the Netherlands
The Clustal Omega servers have requirements about sequence input format. They can read multiple formats [FASTA (Pearson), NBRF/PIR, EMBL/Swiss Prot, GDE, CLUSTAL, and GCG/MSF]. The simplest to use and one we have already got in our P450 sequence files is FASTA format. Each sequence has the first line starting with a > and identifier information. The sequence follows on the next line. Additional sequences are in the same format. I have run >300 sequences through this server and it works fine. I would not recommend doing so many during class since the server will get overloaded.
Considerations when making Clustal Omega alignments.
Some examples given below use human mitochondrial carrier proteins. The human carriers have some sequences with N-terminal extensions (50, 52, 55, 89, 91, 132. and 134). Some have C-terminal extensions (89, 91). These are not useful for making a multiple sequence alignment and they should be trimmed to cut off the extended parts. If you do not, the alignment will be several pages longer and this is a waste of computer time and printer paper. These sequences have already been trimmed.
How to do a multiple sequence alignment
Go to this link. Trimmed human carriers
Select about 5-10 sequences (not all starting at the first sequence) and copy and paste them into one of the Clustal servers. If you use the server here Click on the protein radio button. If you use the Wageningen server you don't have to do that. You should get a result like this:
26 -------------------MGDHAWSFLKDFLAGGVAAAVSKTAVAPIERVKLLLQVQHA 41
30 -------------------MTEQAISFAKDFLAGGIAAAISKTAVAPIERVKLLLQVQHA 41
33 -------------------MTDAAVSFAKDFLAGGVAAAISKTAVAPIERVKLLLQVQHA 41
44 MIQNSRPSLLQPQDVGDTVETLMLHPVIKAFLCGSISGTCSTLLFQPLDLLKTRLQTLQP 60
.. * **.*.::.: *. . *:: :* **. :.
26 SKQISAEKQYKGIIDCVVRIPKEQGFLSFWRGNLANVIRYFPTQALNFAFKDKYKQLFLG 101
30 SKQIAADKQYKGIVDCIVRIPKEQGVLSFWRGNLANVIRYFPTQALNFAFKDKYKQIFLG 101
33 SKQITADKQYKGIIDCVVRIPKEQGVLSFWRGNLANVIRYFPTQALNFAFKDKYKQIFLG 101
44 SDHGSRRVGMLAVLLKVVRTES---LLGLWKGMSPSIVRCVPGVGIYFGTLYSLKQYFLR 117
*.: : .:: :** . .*.:*:* ..::* .* .: *. . ** **
26 GVDRHKQFWRYFAGNLASGGAAGATSLCFVYPLDFARTRLAADVGKGAAQREFHGLGDCI 161
30 GVDKHTQFWRYFAGNLASGGAAGATSLCFVYPLDFARTRLAADVGKSGTEREFRGLGDCL 161
33 GVDKRTQFWRYFAGNLASGGAAGATSLCFVYPLDFARTRLAADVGKSGAEREFRGLGDCL 161
44 GHPPTALESVMLG-----VGSRSVAGVCMS-PITVIKTRYESGK-----Y-GYESIYAAL 165
* :. *: ..:.:*: *: . :** :. :..: .:
26 IKIFKSDGLRGLYQGFNVSVQGIIIYRAAYFGVYDTAKGMLPDPKNVHIFVSWMIAQS-- 219
30 VKITKSDGIRGLYQGFSVSVQGIIIYRAAYFGVYDTAKGMLPDPKNTHIVVSWMIAQT-- 219
33 VKIYKSDGIKGLYQGFNVSVQGIIIYRAAYFGIYDTAKGMLPDPKNTHIVISWMIAQT-- 219
44 RSIYHSEGHRGLFSGLTATLLRDAPFSGIYLMFYNQTKNIVPHDQVDATLIPITNFSCGI 225
.* :*:* :**:.*:..:: : . *: .*: :*.::*. : .:. .
26 -VTAVAGLVSYPFDTVRRRMMMQSGRKGADIMYTGTVDCWRKIAKDEGAKAFFKGAWSNV 278
30 -VTAVAGVVSYPFDTVRRRMMMQSGRKGADIMYTGTVDCWRKIFRDEGGKAFFKGAWSNV 278
33 -VTAVAGLTSYPFDTVRRRMMMQSGRKGTDIMYTGTLDCWRKIARDEGGKAFFKGAWSNV 278
44 FAGILASLVTQPADVIKTHMQLYP------LKFQWVGQAVTLIFKDYGLRGFFQGGIPRA 279
. :*.:.: * *.:: :* : . : : . :. * :* * :.**:*. ...
26 LR-GMGGAFVLVLYDEIKKYV---- 298
30 LR-GMGGAFVLVLYDELKKVI---- 298
33 LR-GMGGAFVLVLYDEIKKYT---- 298
44 LRRTLMAAMAWTVYEEMMAKMGLKS 304
** : .*:. .:*:*:
The numbers on the left are the sequence identifiers. Long identifiers will be truncated. Numbers on the right are # of amino acids. There are parameters that you can set and we need to talk about them. The two types of alignment that the program does are pairwise alignment of two sequences and multiple alignment of two blocks of aligned sequences with each other or a single sequence with a block of aligned sequences. These have separate parameters. The values that you may want to change are the gap opening penalty and the gap extension penalty. The default values have been chosen by trial and error to be pretty good values, but if your alignment has some odd gaps in it or sequences are not lining up as you think they should then do not hesitate to change these values. Higher gap penalies will make gaps less frequent and higher extension penalties will make gaps shorter. The other option you have that can make a significant difference is the matrix used to score the proteins. This can be BLOSUM (BLOcks SUbstitution Matrix) or PAM (Accepted Point Mutations) or two others. BLOSUM is built based on the BLOCKS Database The ID matrix only scores identities, so this is a very simple matrix. In my use of this server, I find PAM to be better than BLOSUM at the mito carrier alignments.
The alignment output that we got was clustal. This can be set to other formats depending on what you want to do with it. [Note: the Wageningen server only produces the Clustal format.] Later, we will be using the alignments to make trees using Phylip, so then you will want to select the Phylip output option. [Note: you can convert the Clustal format to other formats including Phylip in the Seaview sequence editor.] Do a Clustal Omega alignment on any 4 carrier sequences and request the output as PHYLIP format for later use (or use the default Clustal format in the Wageningen ClustalW server). When the result comes back select the text from the number line (one line above the sequence alignment) through to the end of the sequence alignment. DO NOT SELECT ALL THE OTHER OUTPUT ABOVE THE SEQUENCE ALIGNMENT. Paste this in Word, name it Carrier1.aln and save as text only on the desktop or in My Documents.
Evaluating the alignment results
Alignments can tell you several things. If you have made a mistake in assembling a gene, it will often turn up in the alignment. I show one example below.
89 LPYNLAELQRQQSPG-LGRPIWLQIAESAYRFTLGSVAGAVGATAVYPIDLVKTR-MQNQ 357 91 LPFNLAEAQRQKASGDSARPVLLQVAESAYRFGLGSVAGAVGATAVYPIDLVKTR-MQNQ 359 94 ----------------------------------GGIAGLIGVTCVFPIDLAKTR-LQNQ 39 95 ----------------------------------GGVAGLVGVTCVFPIDLAKIR-LQN- 38 132 ------------------------------HMTAGAMAGILEHSVMYPVDSVKTR-MQSL 76 134 ------------------------------HMVAGAVAGILEHCVMYPIDCVKQTRMQSL 61In this example, the bottom sequence is too long by one amino acid. The sequence lines up very well except for the Q in VKQTR. This is the end of an intron (CAG) that was not removed in assembling this gene. Below is another section of the carrier alignment.
106 RPSMASELMPSSRLWSLSYTKLPSSLQSTGKCLLYCNGVLEPYLCPNGARCATWFQDPTR 102 106A RP-----------------------SMASGKCLLYCNGVLEPYLCPNGARCATWFQDPTR 79 106B RPSMASELMPSSRLWS--------LSYTKWKCLLYCNGVLEPYLCPNGARCATWFQDPTR 94In this example, there is alternative splicing. The three sequences are identical except in this region. The gaps are missplaced, since the SMAS should pull back to the other side of the gap on sequence 106A and the LSYTK should also pull back on sequence 106B. This is something that might be fixed by choosing different matrices for scoring. Changing gap penalties will not affect this because the gap lengths will stay the same. It is interesting to note that this error does not occur in the EBI Clustal Omega server. By changing parameters of the CLUSTALW server here I was able to get the correct alignment using the Gonnet scoring matix for the multiple alignment parameters.
55 -MLEGS-PQLNMVG---LFRRIISKEG--IPGLYRGITPNFMKVLPAVGIS-----YVVY 465 57 -VLPEF-EKCLTMRDT-MKYDYGHHGI--RKGLYRGLSLNYIRCIPSQAVA-----FTTY 322 62 -VTGYP-RASIAR--T-LRTIVREEGA--VRGLYKGLSMNWVKGPIAVGIS-----FTTF 295 165 -GSSAS-LVLKRLGFKGVWKGLFARII--MIGTLTALQWFIYDSVKVYFRLPRPPPPEMP 350 167 -GSSAS-LVLKRLGFKGVWKGLFARII--MIGTLTALQWFIYDSVKVYFRLPRPPPPEMP 351In this example, sequence 165 and 167 do not line up with conserved parts of the other sequences. The alignment is fine between the two sequences 165 and 167, so the parameters for pairwise alignments are good. By increasing the gap penalty for multiple alignments to 12 this misalignment was not corrected, but changing to the PAM matrix and gap penalty of 12 did fix this problem in the alignment. (see below)
55 ENMKQTLGVTQK------------------------------------------------ 477 57 ELMKQFFHLN-------------------------------------------------- 332 62 DLMQILLRHLQS------------------------------------------------ 307 165 ESLKKKLGLTQ------------------------------------------------- 361 167 ESLKKKLGLTQ------------------------------------------------- 362
55 MLEGSP-----QLNMVG--LFRRIISKEGIPGLYRGITPNFMKVLPAVGISYVVY 465 57 VLPEFE-----KCLTMRDTMKYDYGHHGIRKGLYRGLSLNYIRCIPSQAVAFTTY 322 62 VTGYPR-----ASIAR--TLRTIVREEGAVRGLYKGLSMNWVKGPIAVGISFTTF 295 165 ----------------KGSSASLVLKRLGFKGVWKGLFARIIMIGTLTALQWFIY 332 167 ----------------KGSSASLVLKRLGFKGVWKGLFARIIMIGTLTALQWFIY 331The parameters for the alignment will never be absolutely right for all regions of a complex alignment. That is why you need to look at the alignment and make adjustments to it based on your ability to see things that the program just does not know about. For example, there may be certain motifs or signature sequences that you know are present in every member of a protein family. If these motifs are not very strongly conserved in their sequence an alignment program can easily fail to line them up. This is what happened in the sequences shown above. As the expert, it is your task to take an alignment given by Clustal Omega and improve it by your expert knowledge. This requires either working with the sequences in a word processor or a special type of program called a sequence editor. The word processor can be used, but it has some limitations. If the sequences are longer than 70 amino acids or nucleotides, then the sequence must be represented as multiple pages. This can be a real irritation if you have to add gaps into the alignment, because the position of all the sequence after the new gaps has to be shifted even if that is over 10 pages of an alignment. Sequence editors are built to keep the sequence in an array and display only one section of it at a time. Addition of gaps is not a problem because the array is just made longer. Also the sequence editor can color code the amino acids in any way you define, This can help identify sections of a sequence that are out of alignment with the other sequences around it. The main problem with sequence editors is they are very very particular about the sequence format of the files you ask them to read. If they ask for Phylip format and even one character is not in that format the file will not be read. This can lead to great frustration. In Phylip format, even a blank space following a line of sequence can throw the program off. It tries to count the blank space as a character and either crashes or tells you the sequences are out of alignment. To avoid this problem, alignments to be read into the programs need to be edited in a word processor to remove all offending blanks at the ends of lines. This is done easily in Word by using the paragraph mark button on the top right side of the tool bar. This reveals were the line returns are placed and shows any blanks at the ends of lines.
55 ENMKQTLGVTQK------------------------------------------------ 477 57 ELMKQFFHLN-------------------------------------------------- 332 62 DLMQILLRHLQS------------------------------------------------ 307 165 DSVKVYFRLPRPPPPEMPESLKKKLGLTQ------------------------------- 361 167 DSVKVYFRLPRPPPPEMPESLKKKLGLTQ------------------------------- 362
Because sequence editors are so graphical, they are usually not written to run on both Macs and PCs. Depending on which system you have you may need to download different programs. The editor we will use on these Windows machines is called SeaView. It was written by Manolo Gouy and it can be downloaded from this link. A gif image of what the editing screen looks like is shown here
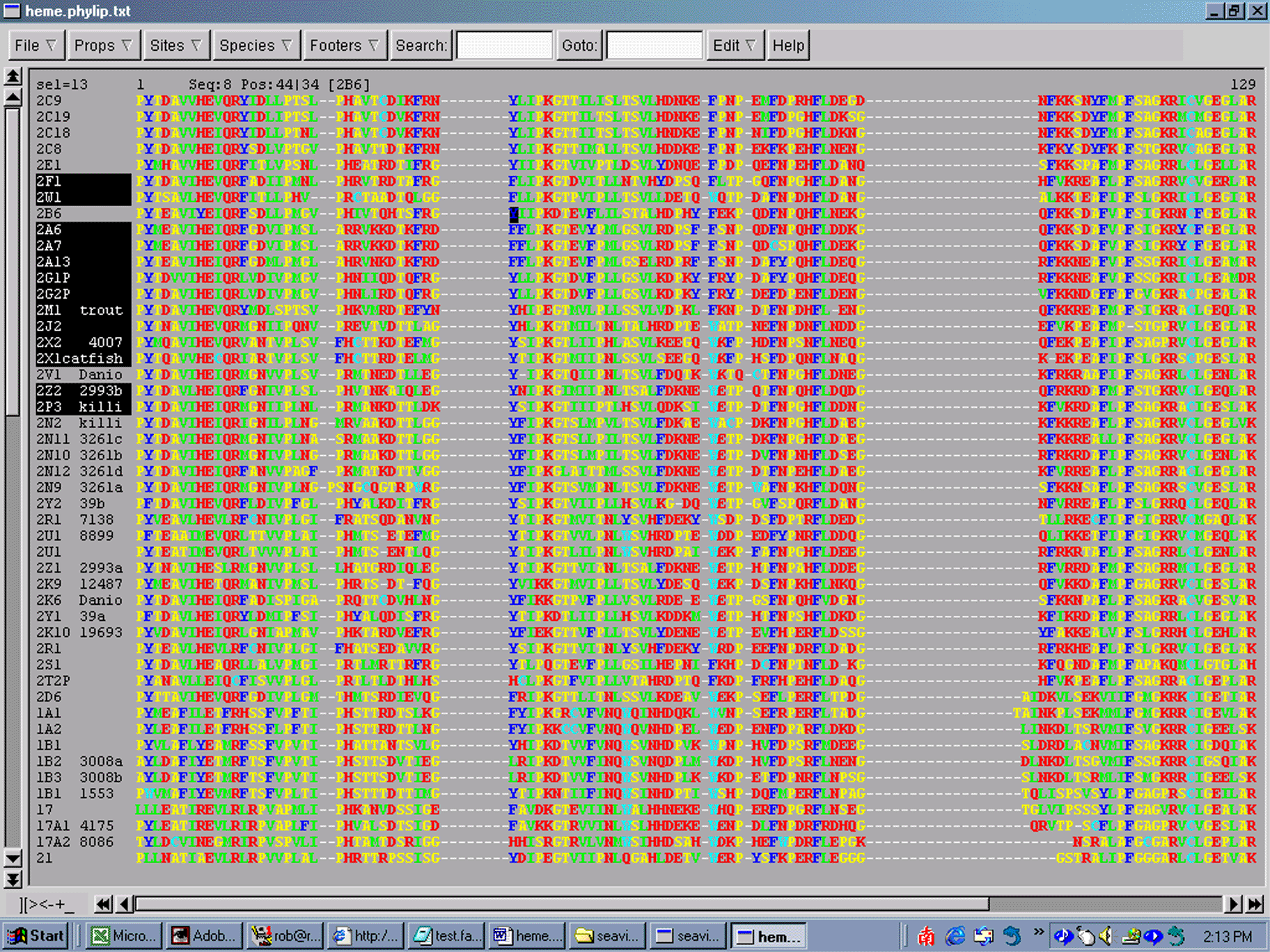{kind=link}
The image shows a section of an alignment of Fugu and human P450s. At amino acids 9-11 is the highly conserved EXXR motif seen in the K-helix of all P450s. If you scroll down the page you will see one exception to this motif. T2TP contains EIQC which violates the EXXR conservation. However this is a pseudogene and it is not functional. Far to the right if you scroll over all the way you will find the heme signature region of P450s. The first sequence has FSAGKRICVG. The signature is FXXGXXXCXG. Notice that the F is missing in one sequence. That is probably an error in this sequence.
Another sequence editor written for Macs is called Se-Al. This one was written by Andrew Rambaut at the Dept. of Zoology at Oxford with support from The Wellcome Trust. It can be downloaded here.
We will use 85 Xenopus P450s as a test file today. This version is in Clustal format. A second version of 83 sequences is in phylip format Xenopus P450s Alignment Phylip
The Phylip format is the one we will actually use, but Clustal format is easier to look at, since all the sequence lines are labeled. MASE format is mentioned by the authors as having lots of room for long names and other descriptive information such as accession numbers, species names etc. This is not included in formats like Phylip where there are only 10 spaces reserved for a name. The authors really like the MASE format for that reason. However, because we will be using Phylip later, we will use Phylip format in this class. I removed two nearly identical sequences from the Xenopus 85 sequence file and fixed an assembly error to make the 83 sequence file.
Please copy the Phylip formatted file and paste it in a word processor and save as Xenopus text only format. You should save it on the desktop or in My Documents. To use the file go to SeaView on the desktop. Start the SeaView program. To open your Xenopus file use the file tab at the top of the SeaView window and select open Phylip. From the dialog box navigate to the desktop or your folder to find your file. At the bottom of the dialog box change files of type Phylip to files of type "all files".
Then click on your filename to open the file in SeaView.
If the file will not open in Seaview, open it in Word and check the format. PHYLIP format has a line with the number of sequences and a space followed by the number of positions in the alignment. These numbers have to be correct. The sequences start with 10 spaces for names. There have to be ten spaces even if they are empty. Character 11 begins the sequence even if it is with -----. There may be spaces in the alignment (often at every 10 amino acids) and there may be spaces between sets of sequences (equivalent to pages in the alignment). There must not be any extra spaces at the end of lines and the names only appear once on the first page. Below is an example. If you copied the Phylip format output from CLUSTAL Omega and saved it as text only this worked for me.
If you use the Wageningen server the output will be Clustal format. Copy the
file beginning with the line that starts Clustal W... to the end.
After pasting in a word file delete the rest of the line after Clustal W.
The SeaView program needs to see Clustal W as the first line of the file to open a
Clustal format file.
Phylip format of a short seq alignment shown earlier
5 115 55 MLEGSP-----QLNMVG--LFRRIISKEGIPGLYRGITPNFMKVLPAVGISYVVY 57 VLPEFE-----KCLTMRDTMKYDYGHHGIRKGLYRGLSLNYIRCIPSQAVAFTTY 62 VTGYPR-----ASIAR--TLRTIVREEGAVRGLYKGLSMNWVKGPIAVGISFTTF 165 ----------------KGSSASLVLKRLGFKGVWKGLFARIIMIGTLTALQWFIY 167 ----------------KGSSASLVLKRLGFKGVWKGLFARIIMIGTLTALQWFIYSeaView example
ENMKQTLGVTQK------------------------------------------------ ELMKQFFHLN-------------------------------------------------- DLMQILLRHLQS------------------------------------------------ DSVKVYFRLPRPPPPEMPESLKKKLGLTQ------------------------------- DSVKVYFRLPRPPPPEMPESLKKKLGLTQ-------------------------------
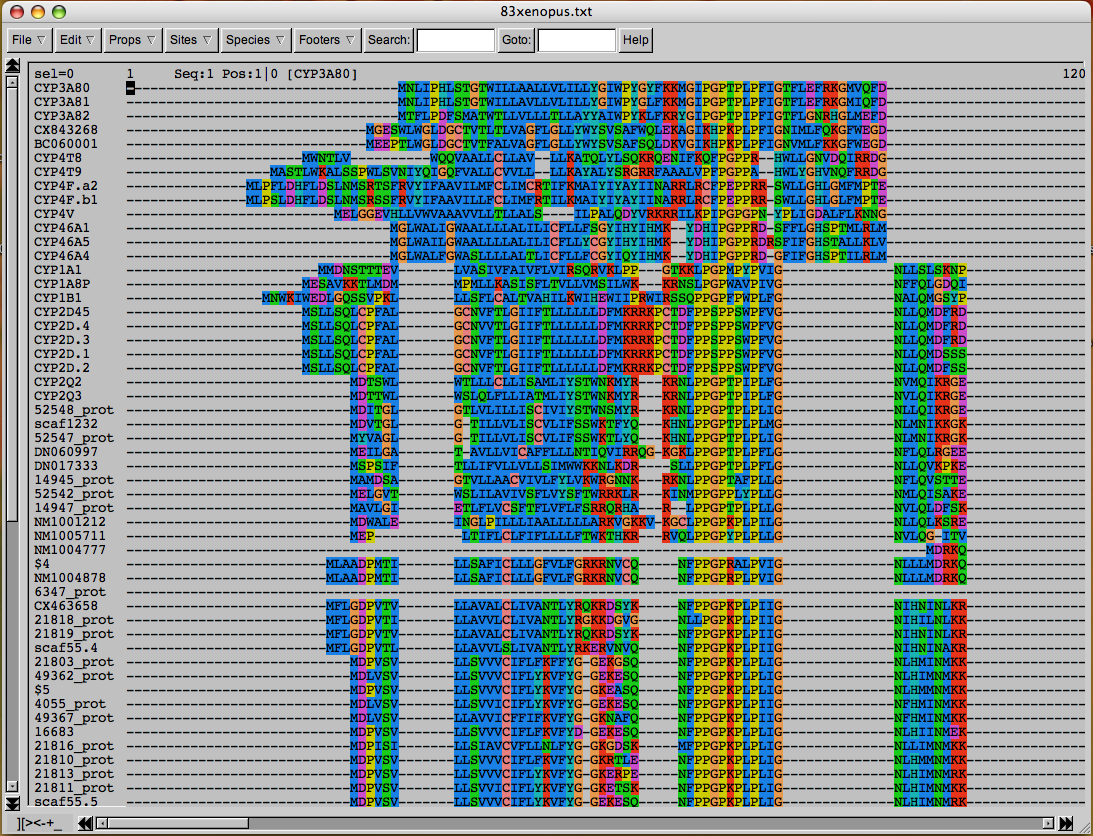
Once the file is open, scroll down using the left side scroll bar and scroll right using the bottom scroll bar. You can see the alignment in full glory. Notice that the names stay on the left as you scroll to the right, so you can always tell what sequences you are looking at. Across the top on the left and right are position counters. These count the alignment position of the left and right edges of the window. These are not amino acid numbers, since the alignment has gaps in it. To get the amino acid number for any amino acid just click on it. The number will be displayed in the same bar as the counters and it will look like this
Seq:26 pos: 126|113 [CYP21 human]
This tells the sequence number, the position in the alignment and the amino acid number from the first position in the alignment (skips counting the dashes) then the name is given in brackets.
To select a sequence click on the name. To select more than one sequence click and drag. To select non-adjacent sequences just click on each one. A second click will deselect the sequence. If you want to clear all your selections to start over, right click on any name (shift click on Macs). It is often necessary to move sequences in an alignment to put them next to one another for comparison. To move a sequence in SeaView, select the sequence by clicking on it. Move your cursor to the location where you want it to go and control click (hold down the control key and click). This will move your selected sequence to the new location. If you want to move a block of sequences select the block by dragging and do the same thing.
This is a sequence editor, so how do you edit the sequences. Place your cursor at the point where you want to change the sequence. You can add dashes or delete dashes to slide the sequences. If you want to add or delete gaps to a set of sequences or even all the sequences, select the sequences as above and then add gaps to one of them. Gaps will be added to all the selected sequences at the same time. To add or remove amino acids you must go to the PROPS menu and check allow seq. edition. If you do not do this you cannot change any of the amino acids. WARNING do not allow sequence edition if you do not want to lose any amino acids. THERE IS NO UNDO BUTTON IN SEAVIEW. Also under the PROPS menu you can allow lower case. I often use lower case to be sequence from a different species that I am using temporarily to fill in a sequence gap. For example, if I have found a Fugu P450 and it is missing the last 20 amino acids, but I have that sequence from medaka (Japanese rice fish), then I can add the medaka sequence in lower case to fill in the missing sequence until I can find it later. After deleting amino acids uncheck the allow seq. edition box to prevent accidental erasure of amino acids.
The edit pull down menu allows you to delete a whole sequence by first selecting it and then choosing the delete command from the edit menu. From this same menu you may delete all columns that contain only gaps. This often happens when you remove sequences or trim sequence ends. There is also a consensus option button that builds a consensus sequence from selected sequences. The dot plot option can let you know if your sequence is in correct alignment with another sequence. If the two sequences are out of alignment the diagonal between the two will not fall on the strict diagonal (in red). This indicates one sequence is shifted relative to the other sequence. You can see this by adding a moderate gap into one sequence and then comparing it to its neighbor in a dot plot.
The color scheme can be altered from the PROPS menu under colors. The default for a protein alignment is protein colors, but there is also inverted colors. This makes the backgound colored and the letters are black. This forms vertical bars of solid color when sequences are conserved. There is a way to make your own color scheme with up to 10 colors. Under customize, you can enter your own color scheme. In the alcolorgroups window type in your amino acids separated by commas and click apply. For example:
In altcolorgroups window type KR,ILMV,AGST,ED,FYW,QNBZ,H,C,P
The color order is fixed and is as follows: red, green, yellow, blue, cyan, magenta, salmon, purple, aquamarine, dark-gray (white is used for dashes). In this example, KR will be red, ILMV will be green, AGST will be yellow, etc. If this file is in the seaview folder when the program is started it will allow you to select alt colors from the PROPS color menu. In this scheme, the positive charges are red, the negative charges are blue, the aromatics are cyan, the aliphatic hydrophobics are green. You can imagine other color schemes that emphasize different properties of the amino acids. If you wanted to color all the charges red and everything else green, use
altcolorgroups = KRED,ILMVAGSTFYWQNBZHCP
Once you have altered the file you can save it and you can print it. You have the option to save as a text file or as a black and white or color PDF. The file dialog box requests a name for the file and it lets you select some options for the PDF, font size and how many amino acids per block. Once the file is saved you can open it in Word or Acrobat and print it. Other programs such as
BOXSHADE. can be used to make a pretty alignment for publication by outlining or shading conserved residues.
More fun with Clustal Omega and JalView.
JalView is a Java based sequence editor that comes built into Clustal W as an output viewing option at the site listed above as
ClustalW server at EBIwith JalView sequence alignment option
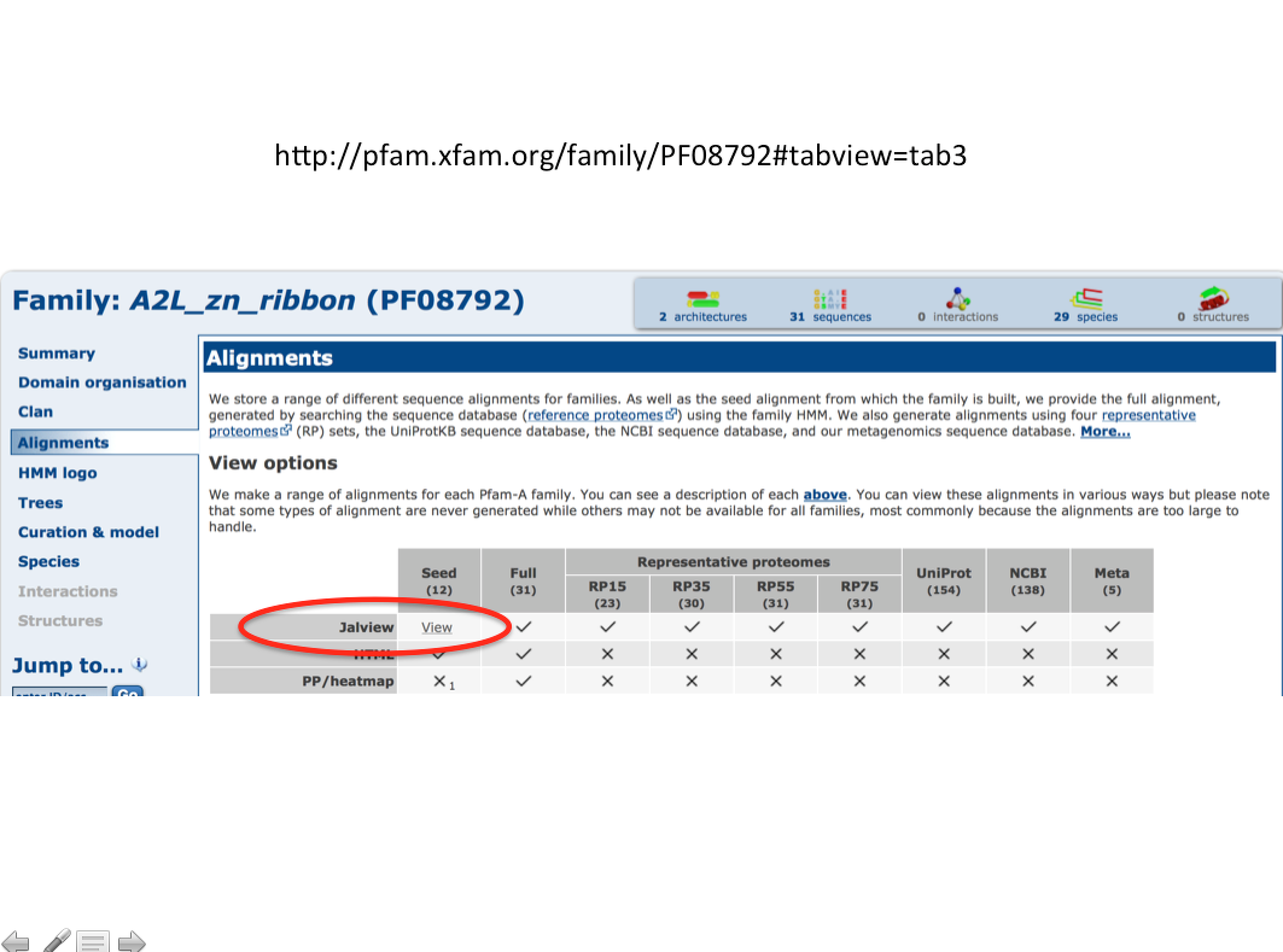
Unfortunately the Clustal Omega site does not use JalView, however Pfam uses it.
You can try out JalView at Pfam, but you will need a current version of Java Runtime on your computer. See the link below to try it.
Pfam example with JalView alignment option
Do a Clustal alignment of all the 49 mouse carriers. Save the output in Phylip format and open in SeaView. Sequence 50b has a long N-terminal extension. Delete this extension and then remove all the gap only columns that this creates (a single command does this removal). This will shorten the alignment. Delete sequence 114a since it is very short. There is one place in the alignment where a gap needs to be added to make the alignment look better. Sequences 50B, 51, 52C, 54 and 56 have an extra glycine G at amino acid 94 in seq 51. Use your cursor to find this amino acid. It should look like this. To introduce a gap to improve the alignment, select all the sequences except the five listed above (you will have to scroll down to do this). Introduce a gap between amino acids G66 and F67 of seq 27 (27 is the sequence name not the sequence position in the alignment). Now deselect all the sequences (right click) and select the five listed above. Introduce a gap in front of M80 of seq 51. It should look like this. This should now have G94 of seq 51 aligning with the gap in all sequences except the five with the extra G. Next move the five sequences to the top of the alignment. It should look like this. This should have tested your editing ability. Prepare a printout with block size 80 characters. Save this as a .pdf file and send it to me.
The following section is out of date.
Go get about 5 or 10 Xenopus P450s in FASTA format and go to this site. Paste in your sequences and do an alignment so you can see the JalView editor. It is really a cool editor. Once the alignment comes back just click on the JalView button in the gray rectangle. The output has the conservation of amino acids as a histogram plot on the bottom. You can mail the alignment back to yourself from the file menu. This editor allows you to slide a sequence block by clicking and dragging. Try it. There is a JalView help button on the EBI Clustal page. This describes in detail all the features of the editor. It is not quite as versatile as the SeaView editor, for example, it does not let you move sequences vertically in the alignment. The grouping feature in this program is a bit confusing. Under the edit pull down menu there is groups and edit groups. In the default settings the alignment opens with all sequences in a group. If you place your cursor somewhere in the sequence and drag to the right it introduces gaps in all sequences at once. If you only want to put a gap in one sequence, you must uncheck the edit groups box in the edit menu. You can also make new groups that will be treated as a set by selecting their names with the mouse and then creating a new group in the edit menu. This is a little confusing until you actually try it.
ASSIGNMENT 3
References
Needleman S.B and Wunsch C.D. (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443-453.
Sellers, P. H. (1974). On the theory and computation of evolutionary distances.
SIAM J. Appl. Math. 26, 787-793.
Smith, T.F. and Waterman, M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol. 147, 195-197.
T. F. Smith, M. S. Waterman, W. M. Fitch (1981)
Comparative biosequence metrics.
Journal of Molecular Evolution, Vol. 18, 38-46.
A tutorial on sequence comparison statistical calculations
Jeanmougin,F., Thompson,J.D., Gouy,M., Higgins,D.G. and Gibson,T.J. (1998)
Multiple sequence alignment with Clustal X. Trends Biochem Sci, 23, 403-5.
Thompson,J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997)
The ClustalX windows interface: flexible strategies for multiple sequence
alignment aided by quality analysis tools. Nucleic Acids Research, 24:4876-4882.
Higgins, D. G., Thompson, J. D. and Gibson, T. J. (1996) Using CLUSTAL for
multiple sequence alignments. Methods Enzymol., 266, 383-402.
Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence
weighting, positions-specific gap penalties and weight matrix choice. Nucleic
Acids Research, 22:4673-4680.
Higgins,D.G., Bleasby,A.J. and Fuchs,R. (1992) CLUSTAL V: improved software for
multiple sequence alignment. CABIOS 8,189-191.
Higgins,D.G. and Sharp,P.M. (1989) Fast and sensitive multiple sequence
alignments on a microcomputer. CABIOS 5,151-153.
Higgins,D.G. and Sharp,P.M. (1988) CLUSTAL: a package for performing multiple
sequence alignment on a microcomputer. Gene 73,237-244.
{kind=link}
{kind=link}
{kind=link}